Mucolipidosis Type II (I-cell Disease): A Rare Condition Resembling Hurler Syndrome: A Case Report
CASE REPORT
Mucolipidosis Type II (I-cell Disease): A Rare Condition
Resembling Hurler Syndrome: A Case Report
S Yang, TW Yeung, HY Lau
Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong
Correspondence: Dr S Yang, Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong
Email: chloe.sy.yeung@gmail.com
Submitted: 30 Jul 2019; Accepted: 30 Aug 2019.
Contributors: SY and TWY designed the study. SY acquired the data. All authors contributed to the analysis of data. SY wrote the article. SY and TWY had critical revisions of the intellectual content of this article. All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have no relevant conflicts of interest to disclose.
Funding/Support: This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics Approval: This study was conducted in accordance with the Declaration of Helsinki. The patient provided verbal informed consent.
INTRODUCTION
Mucolipidosis type II (I-cell disease) is a rare autosomal recessive disorder of lysosomal metabolism with progressive multisystem deterioration that leads to death before or in early childhood. This disease was first described in 1967 by Leroy and DeMars.[1] Children with I-cell disease share many clinical and radiological features with Hurler syndrome although there are distinct differences. Presentation of I-cell disease is earlier with a shorter clinical course, the radiological changes are more profound, and the biochemical features are distinctive. The clinical course of I-cell disease is characterised by progressive failure to thrive and developmental delay. Craniofacial dysmorphic features are similar to those observed in Hurler syndrome although gingival hypertrophy is a distinguishing feature.[2] Previously reported patients with I-cell disease all exhibited craniofacial and/or skeletal abnormalities that were noted on the first day of life although time to diagnosis ranged from 3 weeks to 2.5 years.[3] We report a case of I-cell disease in a patient with a strong family history but no characteristic clinical features present since birth and only radiological abnormality identified on the first day of life.
CASE REPORT
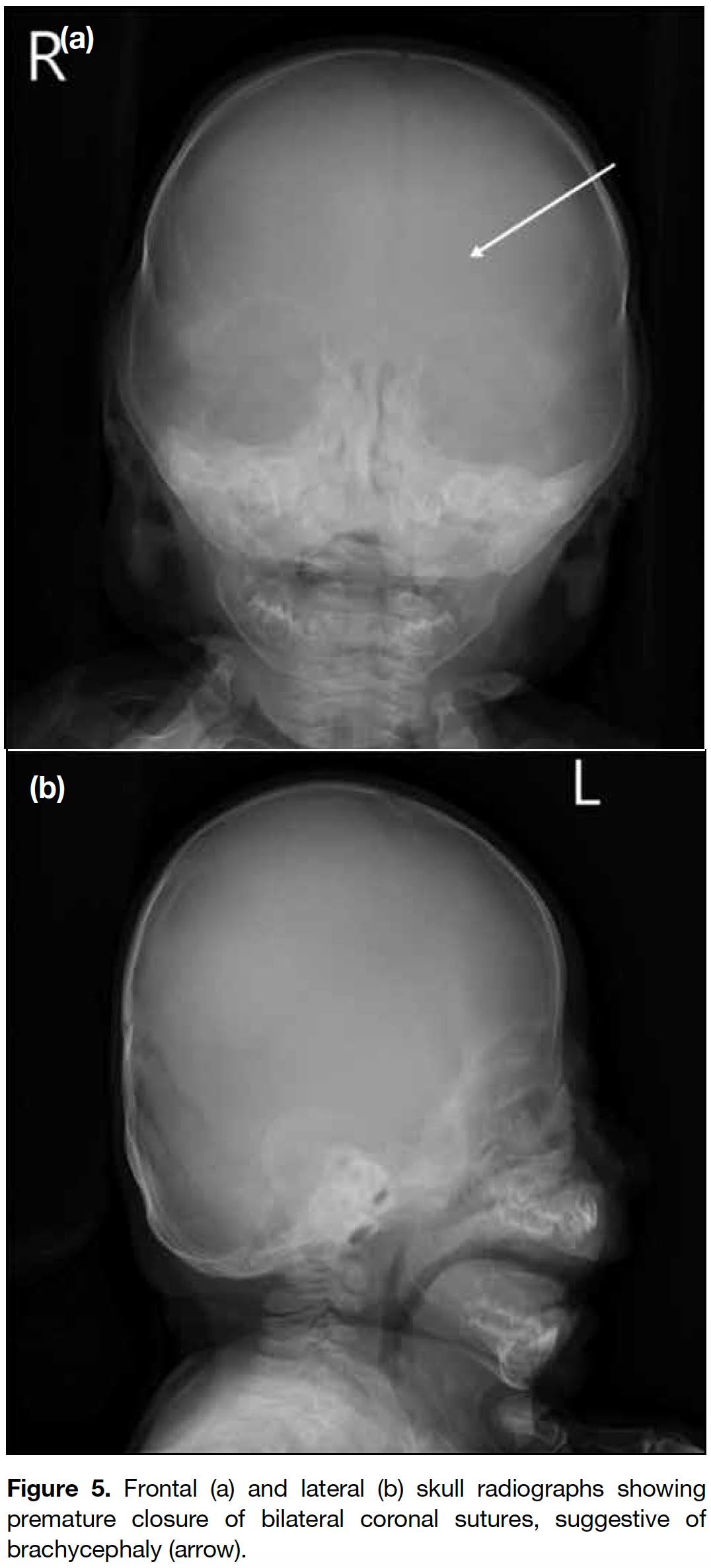
A full term appropriate for gestational age consanguineous Pakistani girl was born by emergency Caesarean section for previous Caesarean section and premature rupture of membrane. Antenatal history was unremarkable. There was a strong family history of I-cell disease. In the extended family, eight children had been diagnosed with the disease and an elder brother died from it at age 7 years. The patient presented with respiratory distress and was diagnosed to have transient tachypnoea of newborn. Initial chest radiograph revealed abnormal widened oar-shaped ribs (Figure 1). A skeletal survey identified simultaneous marked and typical abnormalities of dysostosis multiplex, including oar-shaped ribs, ovoid vertebrae with hypoplasia of anterosuperior corners, thoracic kyphoscoliosis, flaring of the iliac wings with constricted inferior iliac bodies, flattening of acetabular roofs, coarse bony trabeculation, dysplastic capital femoral epiphyses, periosteal cloaking, and rectangular and shortened metacarpals with coned proximal ends (Figures 1 2 3 4 5). Physical examination revealed only insucking of the chest related to respiratory distress. There were no typical clinical features of I-cell disease such as gum hypertrophy, joint contractures or early craniofacial abnormalities. After age 4 months, this patient presented with fever, cough and sputum with repeated admissions for complications of recurrent respiratory infections and consequent respiratory failure.
Figure 1. Supine chest radiograph on the first day of life showing bilateral symmetrical hazy airspace shadows mainly in perihilar region with normal lung volume, suggestive at the time of transient tachypnoea of newborn. Diffuse osteopenia with coarse lacy trabeculae and widened ribs with oar shape were already present.
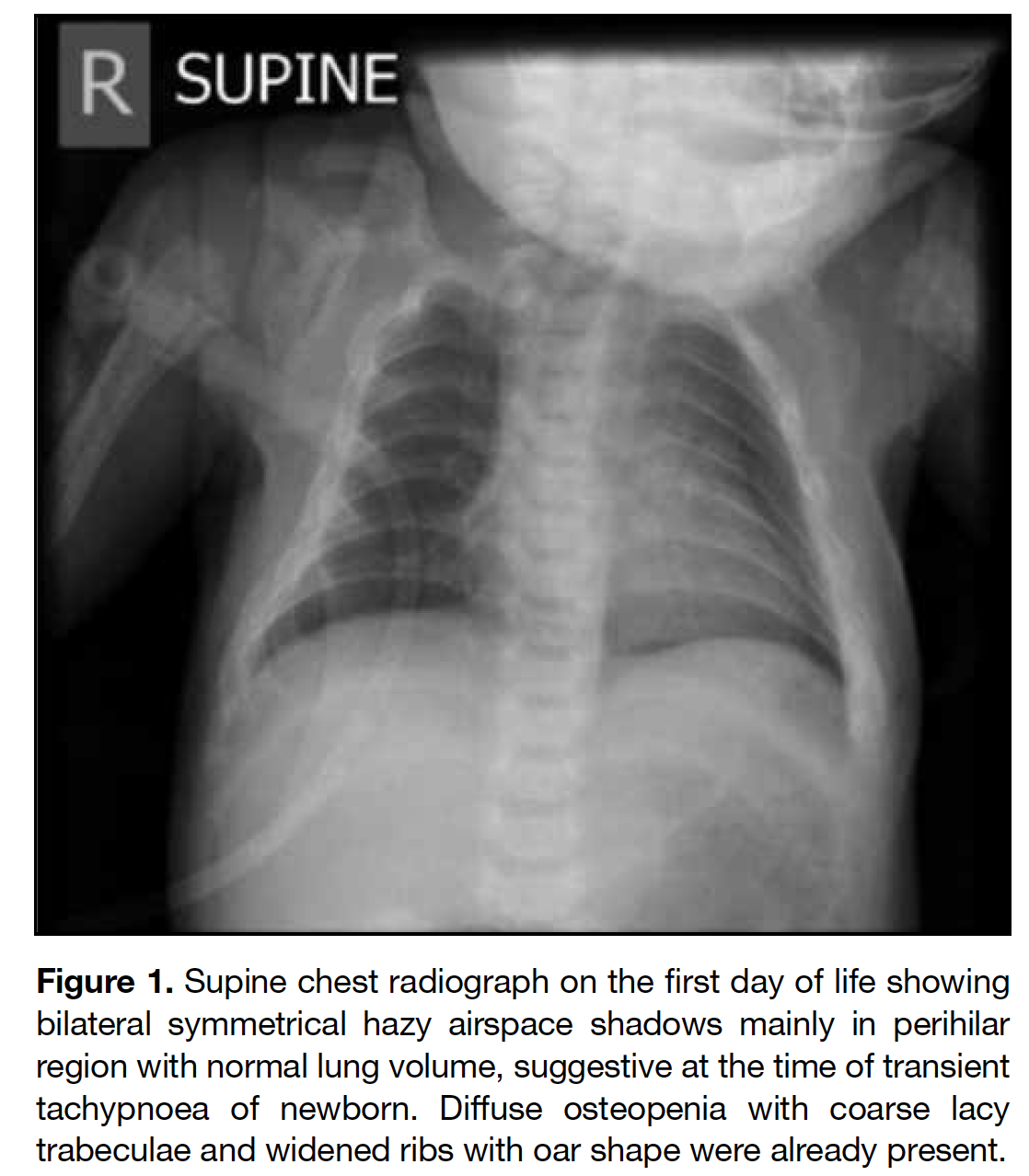
Figure 2. Skeletal survey of bilateral humeri and forearms (a, b), pelvis and femurs (c) showing diffuse periosteal new bone formation at long bones, including bilateral femurs with periosteal cloaking (arrow with tail) and radii (solid arrows). Pelvis and bilateral femurs (c) also demonstrated mild flattening of the acetabular roof of pelvis and constricted inferior iliac body (dashed arrow) with hypoplastic and dysplastic bilateral capital femoral epiphyses (curved arrow).
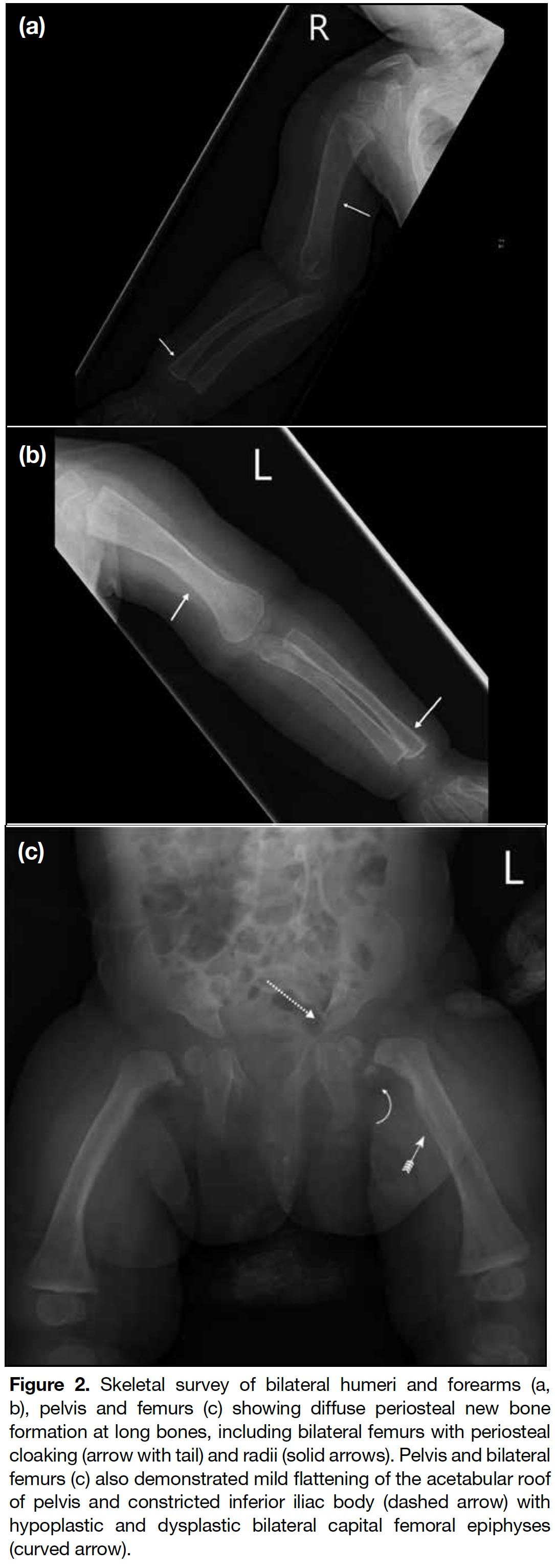
Figure 3. Bilateral hand radiographs (a, b) showing rectangular and shortened metacarpals with coned proximal ends (arrows).
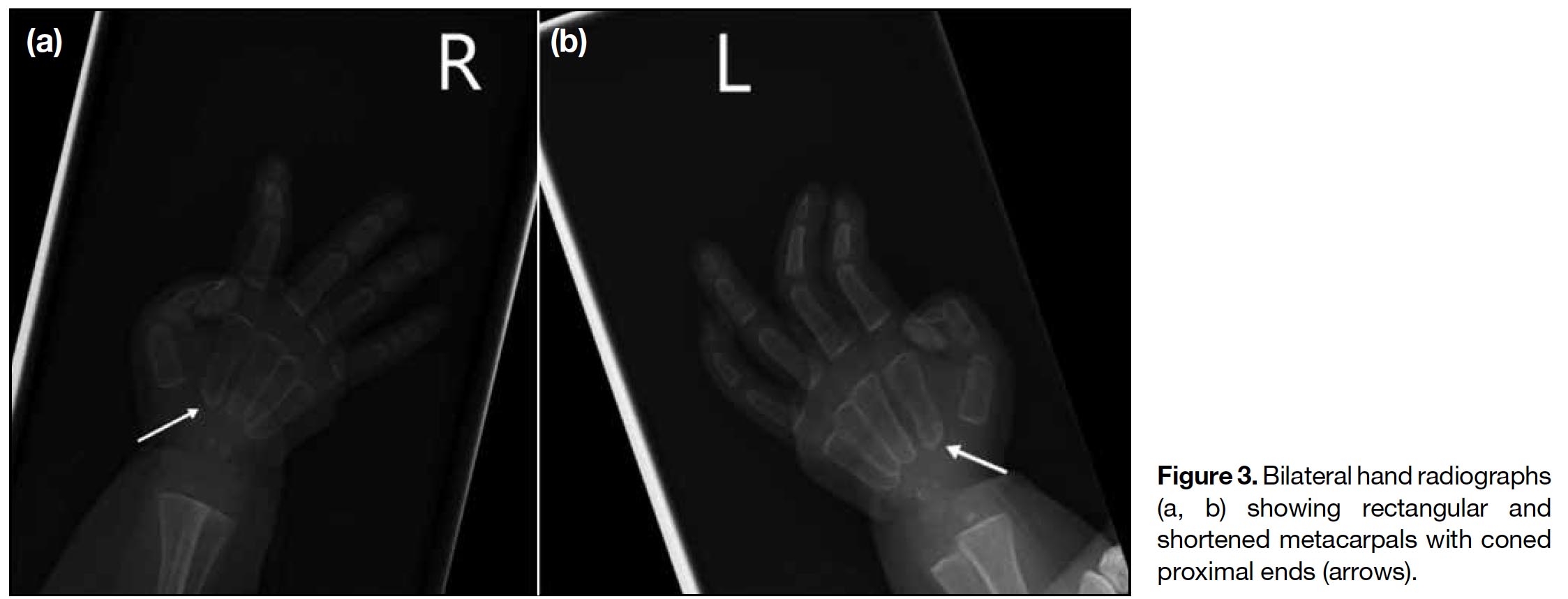
Figure 4. Frontal lumbar spine (a) and lateral spine (b) radiographs showing ovoid and tall vertebrae with hypoplasia of anterosuperior corners (arrow) and thoracic kyphoscoliosis.
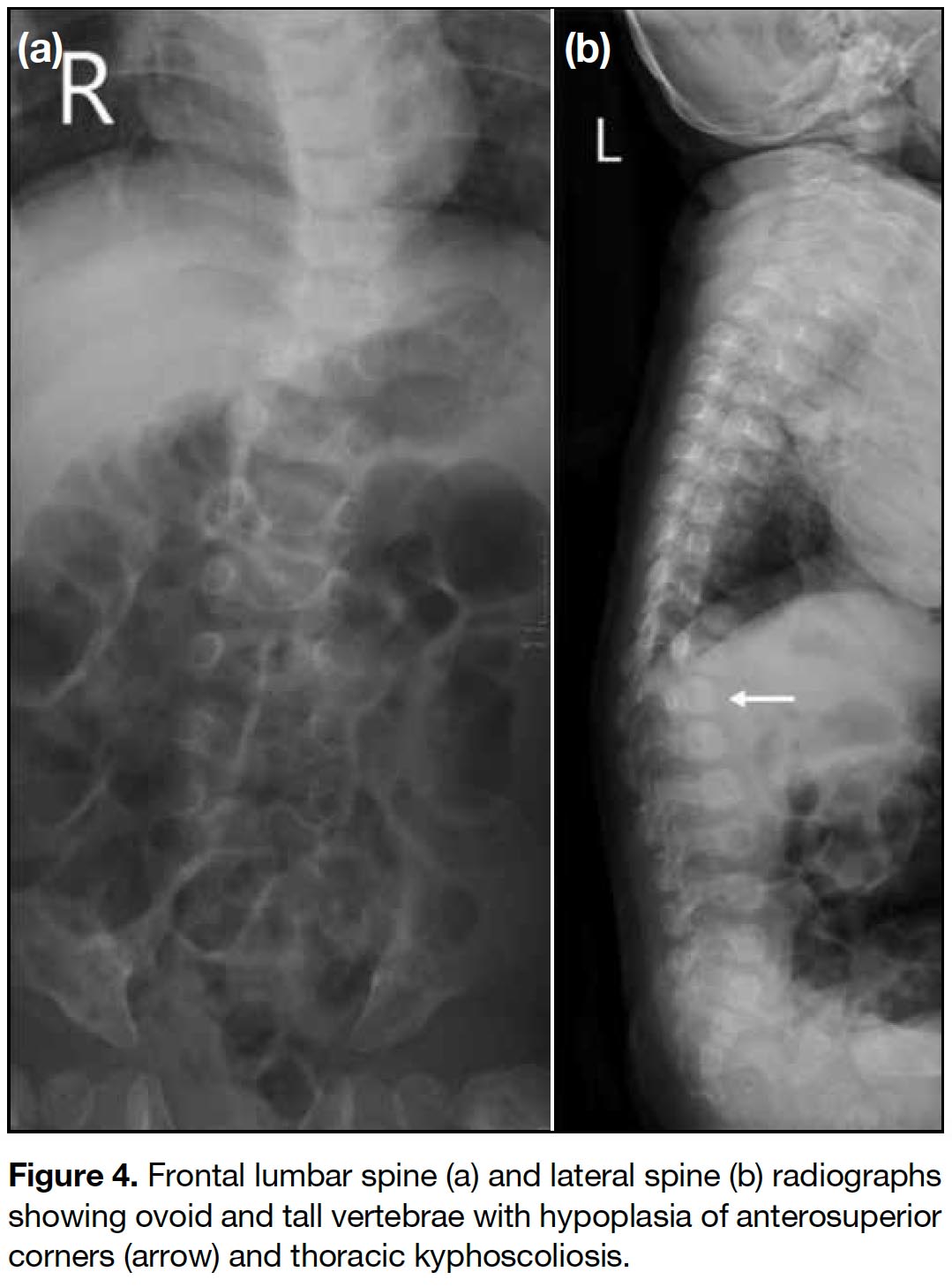
Figure 5. Frontal (a) and lateral (b) skull radiographs showing premature closure of bilateral coronal sutures, suggestive of brachycephaly (arrow).
Urinary metabolic screening and blood genetic testing confirmed the diagnosis of I-cell disease at age 4 months, showing hypersecretion of urinary oligosaccharides and genetic mutation of GNPTAB.
After appropriate counselling, the child’s parents decided not to undergo haematopoietic stem cell transplantation (HSCT). A gastrostomy was performed to facilitate nutritional support due to feeding intolerance. Currently, the patient is receiving optimal supportive management for recurrent respiratory infection and respiratory failure and her nutritional status and growth are being monitored in the outpatient clinic.
DISCUSSION
I-cell disease is a rare inherited lysosomal storage disorder that has no ethnic or gender predilection. The exact prevalence is unknown due to scarce data. Previous studies from different countries estimate a variable prevalence ranging from 1 in 625500 to 1 in 123 500 live births.[4,5] The unique feature of this disease is the presence of numerous phase-dense inclusions in the cytoplasm of fibroblasts. These cells are termed inclusion cells (abbreviated I-cells), and the disease subsequently termed I-cell disease.[6]
Patients with I-cell disease may present with typical physical features at birth, while other features usually become apparent in infancy. Craniofacial abnormalities include coarse facial features, thickened skin, and characteristic hypertrophic gingiva. Skeletal malformations include thoracic deformity, kyphosis, congenital hip dislocation, joint stiffness, clubfeet and deformed long bones. Cardiac involvement is common, with the mitral valve being more frequently affected followed by aortic valve thickening and insufficiency. Occasionally, hepatomegaly and splenomegaly, umbilical and inguinal hernias, and diastasis recti can be present. Neurological deficits such as generalised hypotonia, mild hazy corneas, frequent episodes of otitis media and hoarse voice may be observed.[7] Nonetheless these typical physical features were absent in our patient who exhibited only non-specific thoracic kyphoscoliosis.
Radiological findings of I-cell disease include dysostosis multiplex, a characteristic constellation of radiographic appearance describing the progressive skeletal dysplasia. These changes can be seen in the newborn period and include J-shaped sella turcica; widened ribs at or near the costochondral junctions and narrow dorsal juxtavertebral parts of ribs, namely oar-shaped ribs; vertebral body deformity with superior notching and anteroinferior beaking especially in the lower thoracic and upper-lumbar region; kyphosis and gibbus deformity of the thoracolumbar spine; flaring of the iliac wings with constricted inferior iliac bodies; slanting acetabular roofs and coxa valga; coarse bony trabeculation; hypoplastic/dysplastic capital femoral epiphyses; delayed epiphyseal ossification; bullet-shaped proximal phalanges and proximal pointing of the metacarpals; diaphyseal widening and expansion of tubular bones; metaphyseal cupping and fraying that resemble rickets.[7,8] These features are also present in other type of storage disease, primarily the mucopolysaccharidoses. Some studies have reported findings of cloaking, a transient phenomenon in infants and rarely detectable after the first year, combined with certain radiographic findings in the perinatal/newborn infant period suggestive of I-cell disease. These include talocalcaneal stippling, sacrococcygeal sclerosis, severe generalised vertebral body sclerosis, vertebral body rounding of the lower thoracic/lumbar spine, and rickets/hyperparathyroidism-like changes.[4,9] Other prenatal sonographic abnormalities of I-cell disease have also been reported and include oligohydramnios, intrauterine growth retardation, echogenic cardiac foci, and short femurs and transient alveolomaxillary defect.[3,10] Our patient exhibited most of the imaging features of dysostosis multiplex, including the very characteristic (almost diagnostic) abnormality of cloaking periosteal reaction and characteristic rounding of the thoracic and lumbar vertebrae.[9] In addition, bilateral coronal craniosynostosis was present in our case. This is not commonly seen and only a few cases have been reported. There remains debate about whether it is a component of I-cell disease.[3]
Diagnosis of I-cell disease is based on clinical, radiological, biochemical and molecular findings. Although in most cases typical clinical features are readily apparent at birth, our patient demonstrated no specific abnormalities on physical examination. Therefore, family history and early awareness of the typical radiographic features play a vital role in early diagnosis. The definitive diagnosis is based on biochemical findings that include elevated plasma lysosomal enzyme concentration, deficiency of multiple lysosomal enzymes in cultured fibroblasts, and GNPTAB gene mutations.[11] With the advance of technology, prenatal diagnosis has become possible for those considered at risk.[11,12]
Differential diagnoses of I-cell disease include other lysosomal storage diseases, including the more prevalent Hurler syndrome, GM1-gangliosidosis type 1, infantile galactosialidosis, infantile sialidosis and infantile free sialic acid storage disease. Although patients with I-cell disease share similar clinical and radiological features to mucopolysaccharidoses, it can be distinguished by its earlier onset of clinical signs and more rapidly progressive course leading to death in early childhood, distinctive clinical features, and the absence of mucopolysacchariduria. Three reported unique signs to differentiate I-cell disease from Hurler syndrome include gum hypertrophy, skin infiltration, and the degree of standing height growth retardation.[13] Regardless of these features, biochemical testing can unequivocally distinguish I-cell disease from other conditions.[4]
Prognosis of I-cell disease is poor and ultimately fatal.[3,4] Recurrent respiratory infections and respiratory insufficiency are the common major morbidities (as shown in our case) and have been reported as the main cause of mortality.[8] Currently there is no definitive cure for I-cell disease. Management is mainly supportive and symptomatic: gastrostomy tubes for nutrition and tracheostomy with mechanical support. HSCT has been successfully used only for some forms of lysosomal storage disease, especially Hurler syndrome, before the age of 2 years.[14] Although it has been proposed and attempted in a small number of patients with I-cell disease, it remains experimental with limited data. A recent study revealed that HSCT is inadequate because of the very low survival rate after HSCT and primary disease-related organ damage or infection.[15]
We report a case of I-cell disease in a girl with a strong family history and very typical radiological features after birth. To the best of our knowledge this is the only case reported to be clinically silent in the neonate. Unfortunately, we cannot collect more cases for comparison from her diseased family members as most have either been treated in other countries or have died. Follow-up time is still not long enough to observe the complete natural history and final outcome. Moreover, antenatal investigation of I-cell disease by molecular study was not arranged for this patient. Given the universally fatal prognosis of I-cell disease, especially for at-risk family members, it is best to offer antenatal screening for early detection of the gene mutation. Identification of typical radiological changes in prenatal sonographic study and postnatal radiographs in the appropriate clinical context are paramount in making a prompt diagnosis, helping affected families cope with the disease and outcome.
CONCLUSION
I-cell disease is a rare inherited lysosomal storage disorder with characteristic clinical and radiological features usually present early in childhood and often at birth. The prognosis is grave and there is no cure. Awareness of the typical radiological features in the presence of a family history allows prompt diagnosis and subsequent counselling and management.
REFERENCES
1. DeMars R, Leroy JG. The remarkable cells cultured from a human with Hurler’s syndrome: An approach to visual selection for in vitro genetic studies. In Vitro. 1966;2:107-18. Crossref
2. Leroy JG, DeMars RI. Mutant enzymatic and cytological phenotypes in cultured human fibroblasts. Science. 1967;157:804- 6. Crossref
3. Cathey SS, Leroy JG, Wood T, Eaves K, Simensen RJ, Kudo M, et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010;47:38-48. Crossref
4. Leroy JG, Cathey SS, Friez MJ. GeneReviews® [Internet]: GNPTAB-related disorders Available from: https://www.ncbi.nlm. nih.gov/books/NBK1828/. Accessed 29 Aug 2019.
5. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151-6. Crossref
6. Tondeur M, Vamos-Hurwitz E, Mockel-Pohl S, Dereume JP, Cremer N, Loeb H. Clinical, biochemical, and ultrastructural studies in a case of chondrodystrophy presenting the I-cell phenotype in tissue culture. J Pediatr. 1971;79:366-78. Crossref
7. David-Vizcarra G, Briody J, Ault J, Fietz M, Fletcher J, Savarirayan R, et al. The natural history and osteodystrophy of mucolipidosis types II and III. J Paediatr Child Health. 2010;46:316-22. Crossref
8. Spranger JW, Brill PW, Nishimura G, Superti-Furga A, Unger S. Mucoolipidosis II. In: Spranger JW, Brill PW, Poznanski AK, Spranger, Freed M, Mason P, et al. Bone Dysplasias: An Atlas of Genetic Disorders of Skeletal Development. 2nd ed. New York (NY): Oxford University Press; 2001: 295-9.
9. Lai LM, Lachman RS. Early characteristic radiographic changes in mucolipidosis II. Pediatr Radiol. 2016;46:1713-20. Crossref
10. Chen M, Ke YY, Chang SP, Lee DJ, Chen CH, Ma GC. Prenatal transient alveolomaxillary defect in a case of mucolipidosis II (I-cell disease). Ultrasound Obstet Gynecol. 2010;36:255-6. Crossref
11. Braulke T, Raas-Rothschild A, Kornfeld S. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In: Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell GA, editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York (NY): McGraw-Hill Companies; 2019.
12. Yang M, Cho SY, Park HD, Choi R, Kim YE, Kim J, et al. Clinical, biochemical and molecular characterization of Korean patients with mucolipidosis II/III and successful prenatal diagnosis. Orphanet J Rare Dis. 2017;12:11. Crossref
13. Lemaitre L, Remy J, Farriaux JP, Dhondt JL, Walbaum R. Radiological signs of mucolipidosis II or I-cell disease. A study of nine cases. Pediatr Radiol. 1978;7:97-105. Crossref
14. Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123:1191-207. Crossref
15. LundTC,CatheySS,MillerWP,EapenM,AndreanskyM,Dvorak CC, et al. Outcomes after hematopoietic stem cell transplantation for children with I-cell disease. Biol Blood Marrow Transplant. 2014;20:1847-51. Crossref