Magnetic Resonance Imaging with Spectroscopy Findings in Neonatal Nonketotic Hyperglycinaemia: A Case Report
CASE REPORT
Magnetic Resonance Imaging with Spectroscopy Findings in
Neonatal Nonketotic Hyperglycinaemia: A Case Report
M Cheung, G Ng, W Lam
Department of Radiology, Queen Mary Hospital, Pokfulam, Hong Kong
Correspondence: Dr M Cheung, Department of Radiology, Queen Mary Hospital, Pokfulam, Hong Kong. Email: cm609@ha.org.hk
Submitted: 24 Mar 2019; Accepted: 2 Jul 2019.
Contributors: All authors contributed to the concept and design, acquisition of data, analysis or interpretation of data, drafting of the manuscript, and had critical revision of the manuscript for important intellectual content. All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have no conflicts of interest to disclose.
Funding/Support: This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics Approval: The patient was treated in accordance with the Declaration of Helsinki. Informed consent for all procedures was obtained from his parents.
INTRODUCTION
Nonketotic hyperglycinaemia (NKH) is a rare genetic metabolic disorder with an autosomal recessive inheritance pattern. The condition is caused by an enzyme defect and consequent inability to break down glycine, leading to accumulation within the body’s tissues and fluids, in particular the central nervous system.[1]
Symptom onset can be as early as hours to days after birth. The excessive central nervous system glycine level leads to poor feeding, lethargy, apnoea, progressive encephalopathy, and in cases of classic NKH, death.
Diagnosis of NKH can be challenging and requires a combination of clinical, biochemical, and magnetic resonance (MR) findings.[2,3,4] Genetic testing often confirms the diagnosis. The enzyme system responsible for the breakdown of the amino acid glycine is composed of four proteins, each encoded by different genes. Identifiable genes responsible for the enzyme defect in classic NKH include AMT, GCSH, and GLDC.[5]
There are two forms of NKH: classic and variant. The classic form is further subdivided into mild and severe.
Both severe and mild classic NKH can present in the neonatal period, although the latter can present later in infancy. Initial presentation may be similar for the two forms with hypotonia, lethargy, apnoea, and seizures. However, the clinical course and prognosis of the two forms greatly differ. The severe classic form has a grave prognosis with most patients succumbing or, for the minority of those who survive, severe developmental delay with milestones not reaching those of a typical 6-week-old infant.[6,7] On the contrary, mild classic forms of NKH are associated with variable developmental progress, with patients often being able to walk and develop some motor skills.
Variant forms of NKH are those without mutations in AMT, GCSH, or GLDC. Their clinical course is heterogeneous: some affected individuals achieve normal development, whereas some have severe neurodegeneration with leukodystrophy, cardiomyopathy, and optic atrophy.[8] Similar to the classic mild form of NKH, the outcome is significantly better than for severe classic NKH.[3]
Several papers have discussed the use of neuroimaging in the evaluation of patients with NKH in diagnostic and prognostic evaluations.[3,6,8] This is important for family counselling.
We present the case of an 11-day-old neonate with NKH (genetically confirmed pathogenic variants on GLDC) with an emphasis on MR signal abnormalities, diffusion-weighted imaging (DWI), and proton MR spectroscopy (1H-MRS) findings. We highlight the use of MR findings in facilitating physicians to make an early diagnosis before genetic test results are available and in the prognostic evaluation of the patient, helping guide subsequent clinical management.
CASE REPORT
A term Chinese baby boy of non-consanguineous Chinese parents was born as the first child of the family in a private hospital with negative antenatal history, normal weight, and uneventful birth. He developed poor feeding, lethargy, and decreased responsiveness on day 3 of life. The patient developed recurrent apnoea and progressive encephalopathy on day 4 and was transferred to the neonatal intensive care unit of a tertiary referral centre shortly after emergency intubation and commencement of mechanical ventilation. Clinically, the baby was severely depressed and had almost no spontaneous movements except intermittent hiccups and rare multifocal myoclonic jerks. There was no acidosis, hypoglycaemia, hyperammonia, or ketosis. Complete blood picture, liver function, and renal function were all normal. Blood and cerebrospinal fluid (CSF) cultures were negative.
Continuous electroencephalography monitoring showed burst suppressions with infrequent electrical seizures, not responsive to intravenous pyridoxine but relatively controlled with therapeutic doses of phenobarbitone and phenytoin. Urgent investigation for inborn errors of metabolism showed raised plasma and CSF glycine levels (raised CSF/plasma glycine ratio 0.24) as well as persistently raised lactate and threonine.
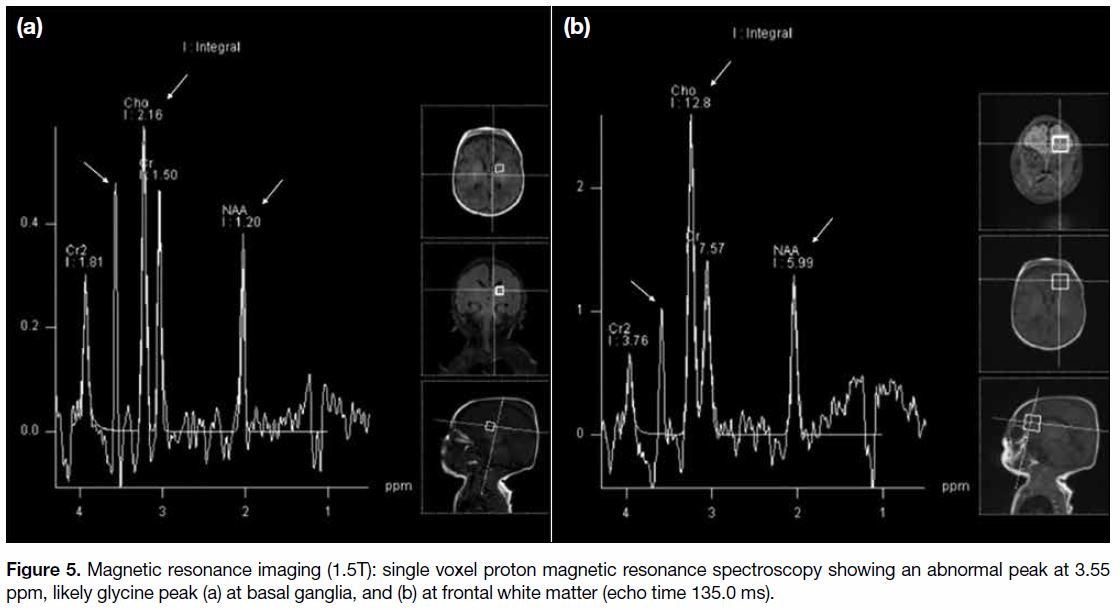
MR and DWI scans of the brain (1.5T) performed at 11 days of life showed loss of normal T2-weighted hypointense signal at bilateral ventrolateral thalami and bilateral symmetrical restricted diffusions with corresponding reduced apparent diffusion coefficient at posterior limbs of internal capsules, lateral thalami, and globus pallidus (Figures < a class="aCrossRefSup" href="#fig1">1 and < a class="aCrossRefSup" href="#fig2">2). There was also brainstem involvement at the cerebral peduncles, ventral midbrain, ventral and dorsal pons, medulla, and bilateral cerebellar peduncles (Figure 3). Hypoplasia with thinning of the corpus callosum was also detected (Figure 4). Single voxel 1H-MRS Figure 5) over the left frontal white matter and left basal ganglia performed at echo time 30.0 ms and 135.0 ms showed an elevated glycine peak at 3.55 ppm. Imaging findings supported the diagnosis of NKH.
Figure 1. Magnetic resonance imaging (1.5T): bilateral symmetrical signal abnormalities in posterior limbs of internal capsules (arrows). (a)
Abnormal hyperintense signal on T2-weighted imaging (repetition time 6000.0 ms, echo time 96.0 ms), (b) restricted diffusion on diffusionweighted
imaging (b1000t/90), and (c) apparent diffusion coefficient map.
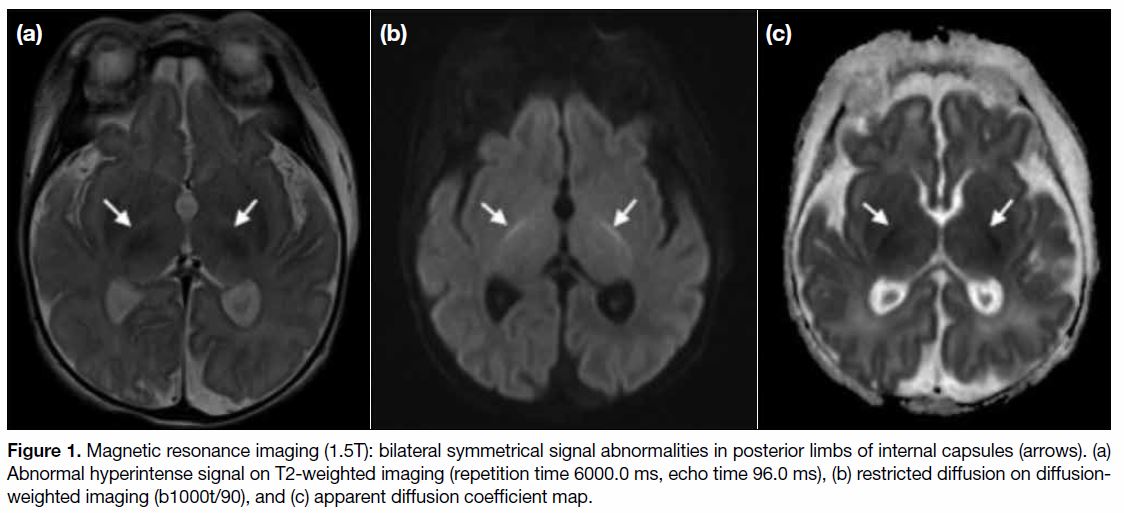
Figure 2. Magnetic resonance imaging (1.5T): bilateral symmetrical signal abnormalities in ventrolateral thalami (arrows). (a) abnormal
hyperintense signal on T2-weighted imaging (repetition time 6000.0 ms, echo time 96.0 ms). (b) restricted diffusion on diffusion-weighted
imaging (b1000t/90), and (c) apparent diffusion coefficient maps.
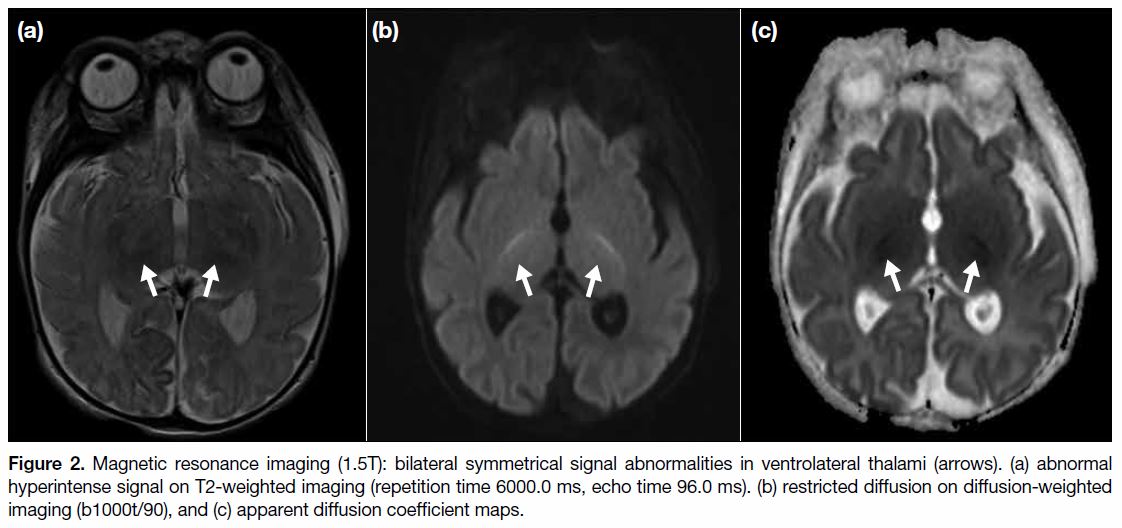
Figure 3. Magnetic resonance imaging (1.5T): multiple axial cuts of the brain stem showing symmetrical restricted signals in cerebral
peduncles and ventral midbrain (white arrows), ventral and dorsal pons (grey arrows), medulla (black arrows) and cerebellar peduncles
(dashed arrows) on (a-d) diffusion-weighted imaging (b1000t/90) and (e-h) apparent diffusion coefficient maps.
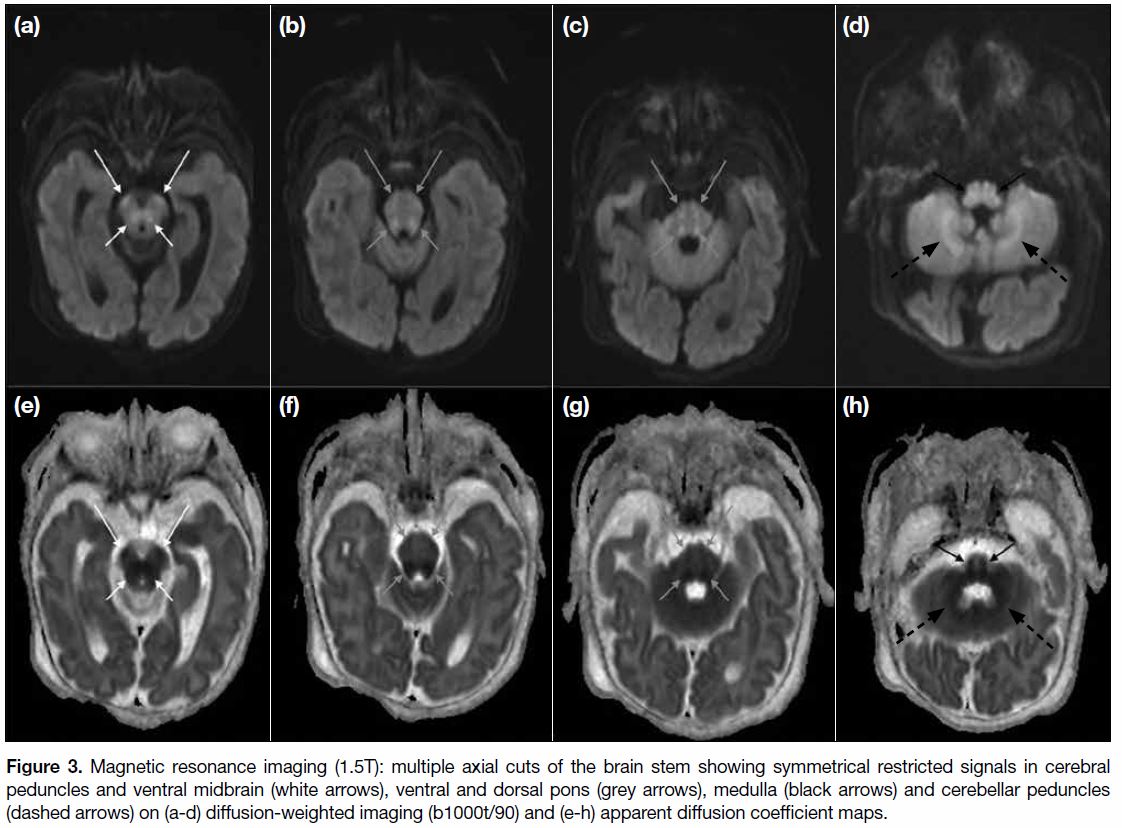
Figure 4. Magnetic resonance imaging (1.5T): sagittal T1-weighted images showing thinning and hypoplasia of the corpus callosum as
marked by arrows (repetition time 450.0 ms, echo time 8.4 ms).
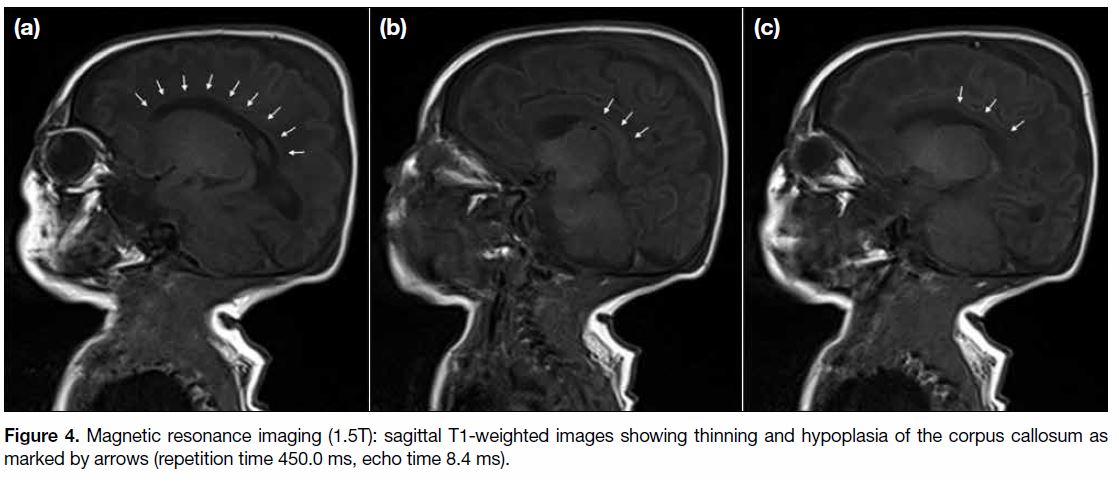
Figure 5. Magnetic resonance imaging (1.5T): single voxel proton magnetic resonance spectroscopy showing an abnormal peak at 3.55
ppm, likely glycine peak (a) at basal ganglia, and (b) at frontal white matter (echo time 135.0 ms).
Sodium benzoate and dextromethorphan were commenced in an attempt to enhance glycine cleavage.
A clinical geneticist was consulted and urgent testing for rapid genetic confirmation was requested.
Despite medical treatment being further optimised, the baby remained very unresponsive with some occasional spontaneous movement, shallow respirations, and minimal improvements in electroencephalography background observed in the subsequent few days. The clinical team additionally sought overseas expert advice regarding baby’s likely diagnosis, prognosis, and appropriateness of considering redirection of care at this stage since the outcome was deemed to be grave. It was agreed that severe classic-type NKH was the most likely diagnosis based on baby’s clinical course, biochemical, and MR findings.
Family counselling was conducted on multiple occasions. The family expressed reluctance to accept a severely disabled child regardless of the underlying disease cause and explicitly opted for active withdrawal of life support in view of the grave prognosis. Psychological and bereavement support was provided to the family. The family’s decision for redirection of care and active withdrawal was reviewed on multiple occasions. A clinical psychologist assessed the parents’ understanding and fitness to give informed consent before they arrived at the final decision of active withdrawal. The baby was electively extubated and he passed away on day 18 of life. Post-mortem biopsies were saved, and confirmatory genetic analysis identified a rare missense variant in exon 21 and a deletion in exon 3 on GLDC. No other relevant pathogenic variant was identified by whole exome sequencing analysis.
DISCUSSION
NKH is a rare disorder of inborn error of glycine metabolism caused by a genetic defect in the breakdown of the amino acid leading to accumulation of toxic levels in the blood, brain, CSF, organs, and tissues. In severe classic forms, it is often fatal.
In the absence of acidosis, hypoglycaemia, hyperammonia, ketosis and sepsis, urgent inborn errors of metabolism investigations should be sought. A highly elevated glycine level in the CSF, plasma and CSF/plasma glycine ratio are the first clues to a diagnosis of NKH.
The underlying histopathology of NKH is vacuolating myelinopathy where myelinated areas undergo vacuolation, demyelination, and astrocytosis.[9] Many metabolic diseases, mitochondrial diseases and intoxications can cause vacuolating myelinopathy, thus MR studies of these neonates may all produce a similar pattern of signal abnormalities in areas that are undergoing active myelination at the time.[10]
Previous studies have shown abnormal T2-weighted hyperintensities with restricted diffusion in patients with NKH in the ascending and descending tracts of the brain stem, posterior limbs of the internal capsules, corticospinal tracts, dorsal midbrain, dorsal pons, cerebral and cerebellar peduncles, as well as hypoplasia of the corpus callosum.[10,11] In our patient, the sites of abnormal signal intensity were similarly confined to the white matter tracts that are myelinated at birth, namely dorsal pons, midbrain, cerebellar white matter, posterior limb of the internal capsule, lateral thalamus, and globus pallidus, consistent with vacuolating myelinopathy. Hypoxic ischaemic encephalopathy of a term neonate may share similar signal abnormalities at the basal ganglia and thalamus. However, signal abnormalities would also be additionally seen at the perirolandic cortex and signal abnormalities at the brainstem and cerebellar peduncles are not recognised features.
The different forms of NKH (e.g. classic severe, mild, variant) may present very similarly in the initial clinical setting, but all carry a very different prognosis and outcome, with the classic severe type carrying the worse outcome and grave prognosis. As initial clinical symptoms in patients with variant and mild forms can be similar to classic severe forms, it may present difficulties to the physician in determining the course of the disease and prognosis and in family counselling after the initial diagnosis.
Previous studies have shown the use of 1H-MRS in identifying glycine peaks at 3.55 ppm, and in particular neonates with NKH.[12,13] Swanson et al[7] proposed biochemical and molecular predictors that could help to predict the severity of the disorder, based on genotype, high CSF glycine levels, presence of brain malformations, and age of onset. They found that high levels of glycine in CSF usually predicted severe NKH, whereas low CSF/plasma glycine ratios were more often associated with the mild form. Thus, in addition to conventional MR and DWI, the use of 1H-MRS in identifying the presence of an elevated glycine peak can also help in suggesting a severe classic form of NKH.
In our patient we also performed additional 1H-MRS due to clinical suspicion, revealing an abnormal glycine peak at 3.55 ppm that supported an early diagnosis of NKH. The combination of clinical, biochemical, and MR findings with DWI and 1H-MRS suggested severe classic-type NKH as the most likely diagnosis. It facilitated physicians in the early diagnosis of neonatal NKH, providing prognostic information about the expected phenotypic course and subsequent management plan and family counselling prior to confirmatory genetic testing.
CONCLUSION
This case illustrates the classic clinical presentation and typical imaging findings in a neonate with NKH. It highlights the role of 1H-MRS in addition to conventional MR and DWI in early detection of NKH. A constellation of particular imaging findings together with glycine peak at 3.55 ppm on 1H-MRS carries diagnostic and prognostic implications. The recognition of classic severe, mild, and variant NKH is important. Physicians should take into account such parameters together with other clinical and biochemical assessments to better direct therapy and provide families with prognostic information about the expected phenotypic course.
REFERENCES
1. Nakai T, Nakagawa N, Maoka N, Masui R, Kuramitsu S, Kamiya N. Structure of P-protein of the glycine cleavage system: implications for nonketotic hyperglycinemia. EMBO J. 2005;24:1523-36. Crossref
2. Lang TF, Parr JR, Matthews EE, Gray RG, Bonham JR, Kay JD. Practical difficulties in the diagnosis of transient non-ketotic hyperglycinaemia. Dev Med Child Neurol. 2008;50:157-9. Crossref
3. Dinopoulos A, Matsubara Y, Kure S. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab. 2005;86:61-9. Crossref
4. Boneh A, Allan S, Mendelson D, Spriggs M, Gillam LH, Korman SH. Clinical, ethical and legal considerations in the treatment of newborns with non-ketotic hyperglycinaemia. Mol Genet Metab 2008;94:143-7. Crossref
5. Coughlin CR 2nd, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodríguez-Pombo P, et al. The genetic basis of classical nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med. 2017;19:104-11. Crossref
6. Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35:253-61. Crossref
7. Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, Bjoraker KJ, Spector EB, et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol. 2015;78:606-18. Crossref
8. Baker PR 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, et al. Variant non-ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2014;137(Pt 2):366-79. Crossref
9. van der Knaap MS, Valk J. Magnetic resonance of myelin, myelination, and myelin disorders. New Y ork: Springer-V erlag; 1995: 209-10. Crossref
10. Khong PL, Lam BC, Chung BH, Wong KY, Ooi GC. Diffusion- weighted MR imaging in neonatal nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 2003;24:1181-3.
11. Mourmans J, Majoie CB, Barth PG, Duran M, Akkerman EM, Poll-The BT. Sequential MR imaging changes in nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 2006;27:208-11.
12. Heindel W, Kugel H, Roth B. Noninvasive detection of increased glycine content by proton MR spectroscopy in the brains of two infants with nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 1993;14:629-35.
13. Hattingen E, Lanfermann H, Quick J, Franz K, Zanella FE, Pilatus U. 1H MR spectroscopic imaging with short and long echo time to discriminate glycine in glial tumours. MAGMA. 2009;22:33- 41. Crossref