Familial Haemophagocytic Lymphohistiocytosis: Spectrum of Neuroimaging Findings in Three Siblings
PICTORIAL ESSAY
Familial Haemophagocytic Lymphohistiocytosis: Spectrum of
Neuroimaging Findings in Three Siblings
BYW Li, TW Yeung, HY Lau
Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong
Correspondence: Dr BYW Li, Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong. Email: birgittali@gmail.com
Submitted: 20 Nov 2018; Accepted: 31 Dec 2018.
Contributors: All authors contributed to the concept of study, acquisition and analysis of data, drafting of the manuscript, and had critical revision
of the manuscript for important intellectual content. All authors had full access to the data, contributed to the study, approved the final version
for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have disclosed no conflicts of interest.
Funding/Support: This pictorial essay received no specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Ethics Approval: This study was approved by the New Territories West Cluster Research Ethics Committee (Ref NTWC/REC/20001).
BACKGROUND FAMILY HISTORY
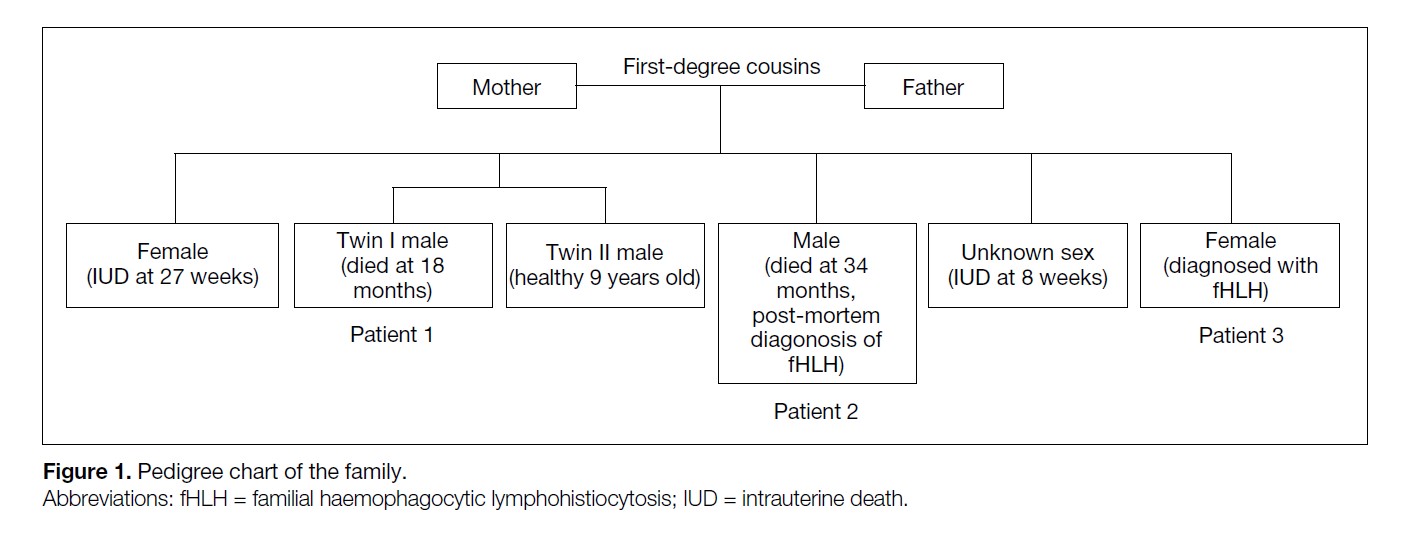
A consanguineous Pakistani couple presented with a
history of recurrent miscarriages and infant deaths (Figure 1). Their first pregnancy ended with intrauterine death at
27 weeks of gestation. The second pregnancy produced
twins: twin I died at age 18 months and twin II was male
and currently healthy with normal development. The
third pregnancy produced a male who presented with
status epilepticus and global developmental delay at age
2 years. He developed persistent breakthrough seizures
despite being prescribed long-term anticonvulsants
and died at age 34 months after developing multiorgan
failure. The fourth pregnancy ended with miscarriage
at 8 weeks of gestation. The fifth pregnancy produced
a female who was clinically followed up from birth
due to a family history of suspected neurodegenerative
disease. This girl was found to have developmental
delay at age 12 months and became epileptic. She was
subsequently diagnosed with familial haemophagocytic
lymphohistiocytosis (fHLH) following whole exome
sequencing that detected a homozygous missense
mutation in the PRF1 (perforin) gene. Post-mortem
genetic sequencing subsequently identified the same
mutation in the deceased elder brother from the third
pregnancy.
Figure 1. Pedigree chart of the family.
IMAGING FINDINGS
The computed tomography and magnetic resonance
imaging (MRI) brain findings of the three affected
siblings are reviewed, two of whom were diagnosed with
fHLH and the third who died of suspected complications
of the same disease. A review of the imaging findings for
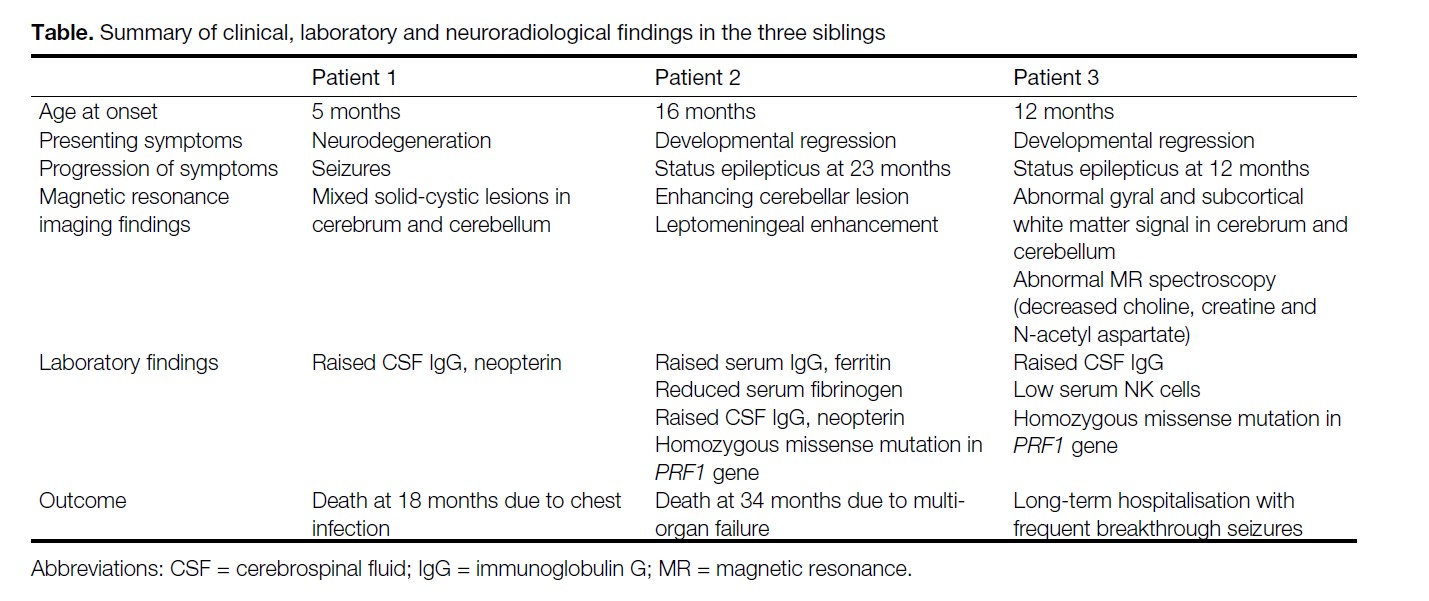
the three patients is summarised in the Table.
Table. Summary of clinical, laboratory and neuroradiological findings in the three siblings.
Patient 1
The first affected sibling was twin I from the second
pregnancy who was born at term following an
unremarkable perinatal history. He presented with signs
and symptoms of neurodegeneration at age 5 months.
Laboratory investigations revealed a salient finding of
elevated serum neopterin and immunoglobulin G.
The initial MRI scan showed an infiltrative heterogeneous
high-intensity T1 signal involving bilateral deep grey
matter (basal ganglia, thalamus), bilateral cerebral
(corona radiata, centrum semiovale) and cerebellar white
matter and brainstem (midbrain and pons).
Differentials at that time included malignancy such as
malignant brainstem glioma, primitive neuroectodermal
tumours, and atypical rhabdoid teratoid tumour.
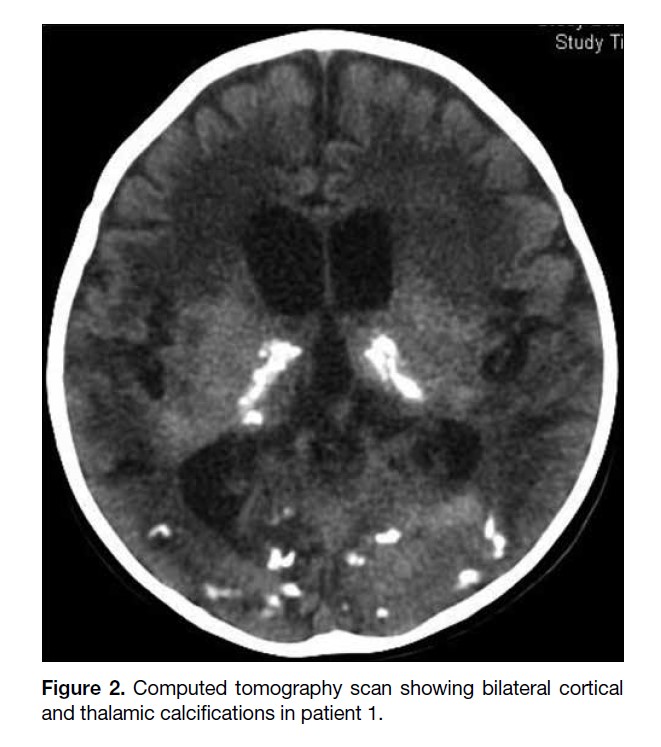
Computed tomography brain studies demonstrated
progressive increase in bilateral cortical and thalamic
calcifications (Figure 2).
Figure 2. Computed tomography scan showing bilateral cortical
and thalamic calcifications in patient 1.
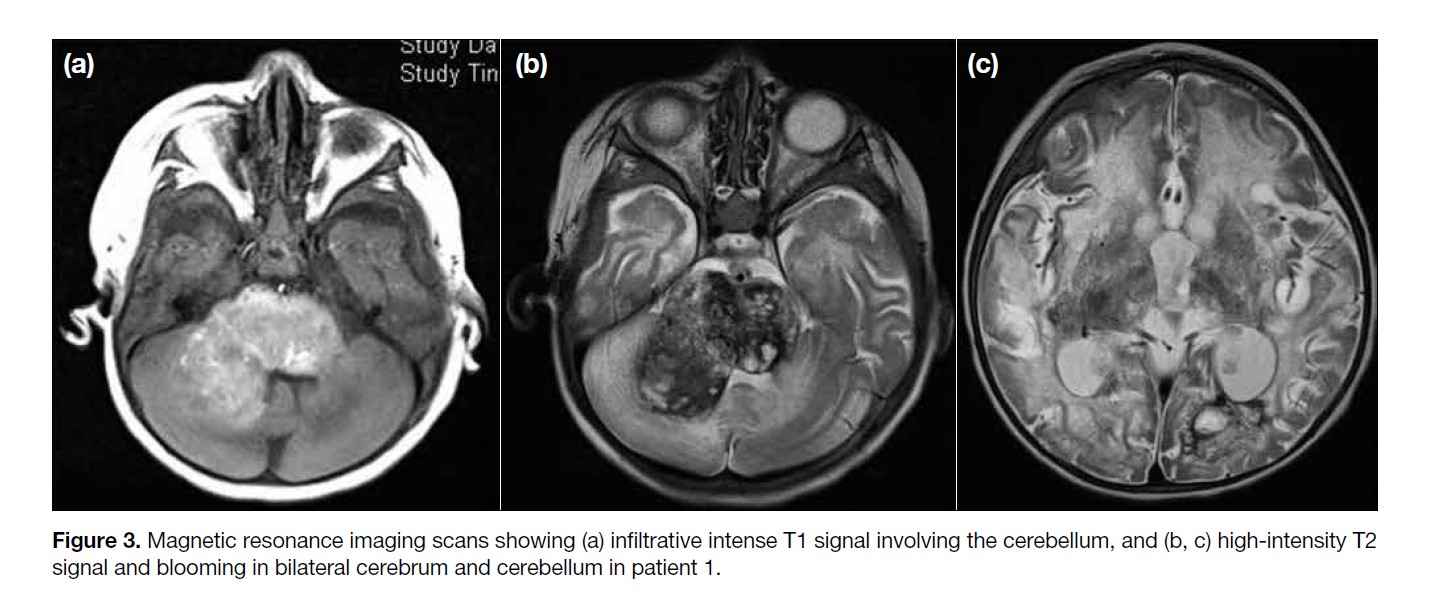
Subsequent MRI scan a month later showed enlargement
of the high-intensity T1 signal lesion in bilateral deep
grey matter, cerebral white matter and brainstem (Figure
3a), and an extensive high-intensity T2 signal in bilateral
cerebral white matter and blooming artefacts suggestive
of calcifications and haemosiderin deposition (Figure 3b and c).
 Magnetic resonance spectroscopy performed in one of
our patients also showed metabolic changes correlating
with neuroimaging findings, with markedly reduced
choline, creatine, and N-acetyl aspartate suggesting
decreased neural density and gliosis representing the end
result of tissue destruction.[9] Focal areas of lactate peak
were suggestive of anaerobic metabolism.
Magnetic resonance spectroscopy performed in one of
our patients also showed metabolic changes correlating
with neuroimaging findings, with markedly reduced
choline, creatine, and N-acetyl aspartate suggesting
decreased neural density and gliosis representing the end
result of tissue destruction.[9] Focal areas of lactate peak
were suggestive of anaerobic metabolism.
Figure 3. Magnetic resonance imaging scans showing (a) infiltrative intense T1 signal involving the cerebellum, and (b, c) high-intensity T2
signal and blooming in bilateral cerebrum and cerebellum in patient 1.
Patient 2
The second case was the male child from the third
pregnancy who developed developmental regression
at age 16 months and was admitted to hospital at age
23 months after presenting with status epilepticus.
Abnormal laboratory tests showed elevated levels of
immunoglobulin G and neopterin in the cerebrospinal fluid
(CSF). Elevated ferritin, triglyceride, immunoglobulin
G and reduced fibrinogen blood serum levels were also
evident. Post-mortem exome sequencing of the patient’s
DNA revealed a PRF1 genetic mutation.
Initial MRI brain showed restricted diffusion at the deep
cortical region of the cerebrum suggestive of hypoxic
ischaemic brain insult. There was also a rim-enhancing
right cerebellar lesion. An infective or inflammatory
process or neoplasm was initially considered.
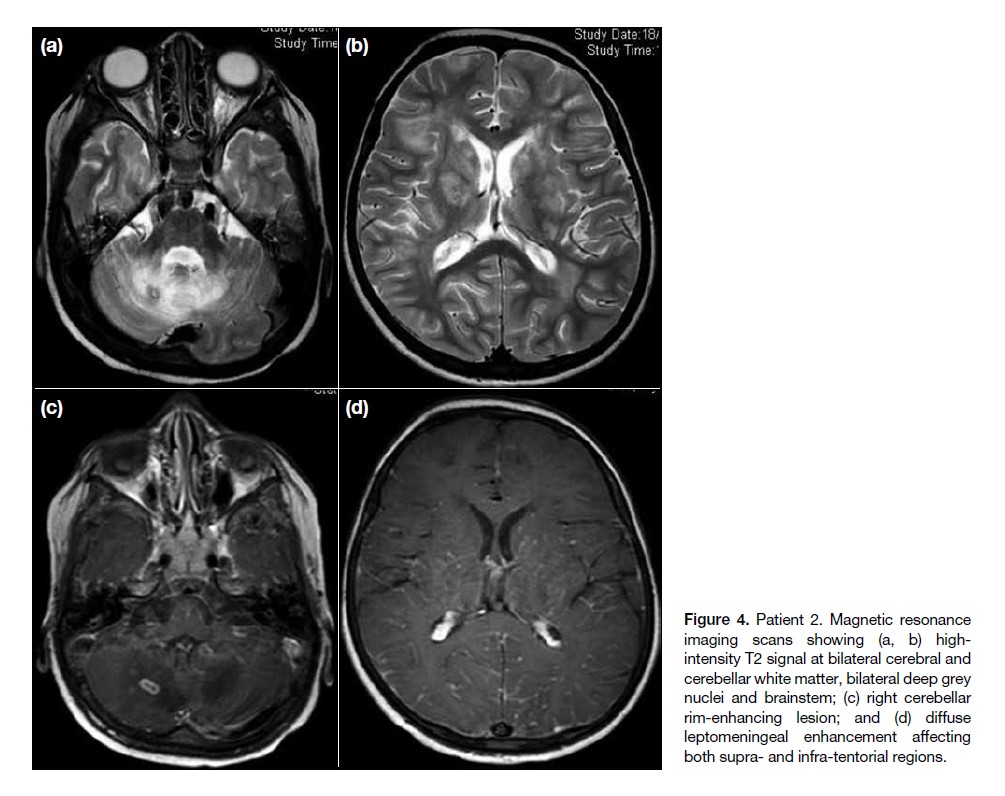
Serial brain MRI scans showed interval enlargement
of the right cerebellar lesion (Figure 4c) and increasing
high-intensity T2 signal in bilateral cerebral and
cerebellar white matter as well as bilateral deep grey
nuclei and brainstem (Figure 4a and b). There was also
development of diffuse leptomeningeal enhancement
(Figure 4d).
Figure 4. Patient 2. Magnetic resonance
imaging scans showing (a, b) highintensity
T2 signal at bilateral cerebral and
cerebellar white matter, bilateral deep grey
nuclei and brainstem; (c) right cerebellar
rim-enhancing lesion; and (d) diffuse
leptomeningeal enhancement affecting
both supra- and infra-tentorial regions.
Patient 3
The female child from the fifth pregnancy presented with
convulsions and status epilepticus at age 12 months.
Elevated serum immunoglobulin G was detected.
Subsequently, whole exome sequencing detected a
homozygous missense mutation at the PRF1 gene.
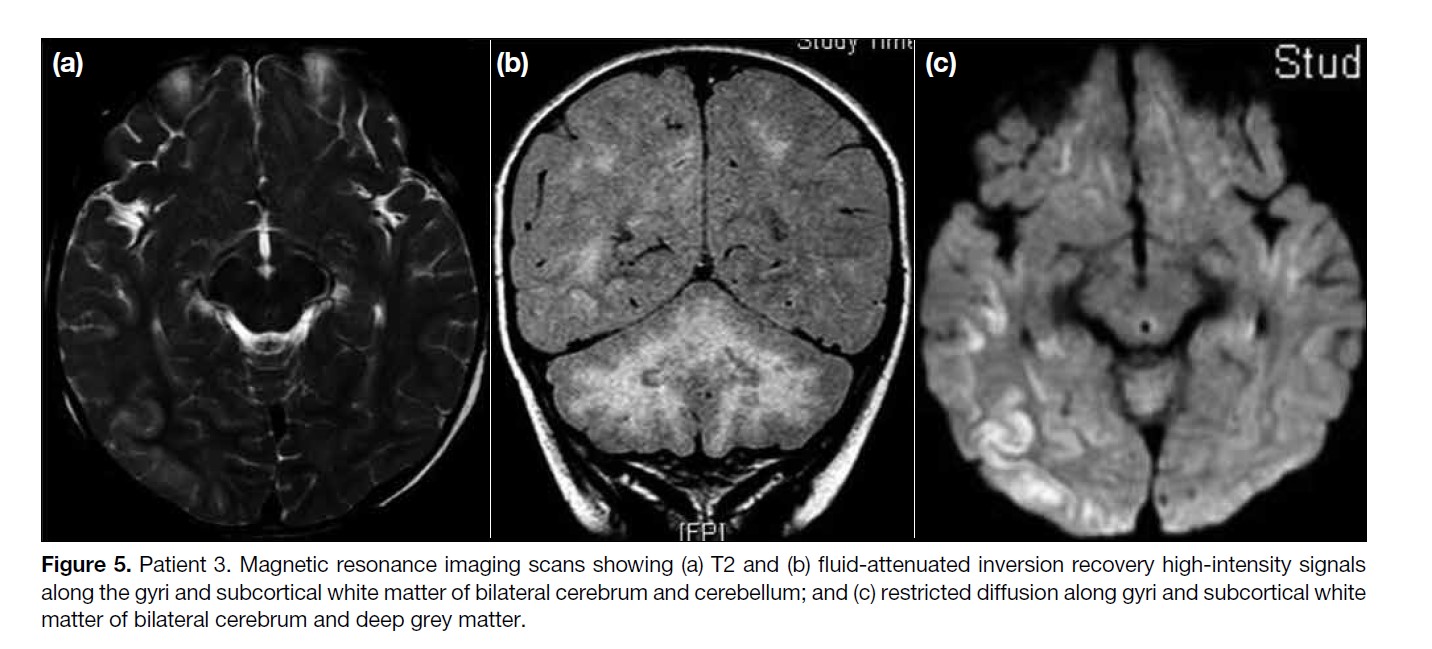
Initial MRI scan showed patchy confluent high-intensity
T2/fluid-attenuated inversion recovery signals along the
gyri and subcortical white matter of bilateral cerebral
and cerebellar hemispheres (Figure 5a and b). There was
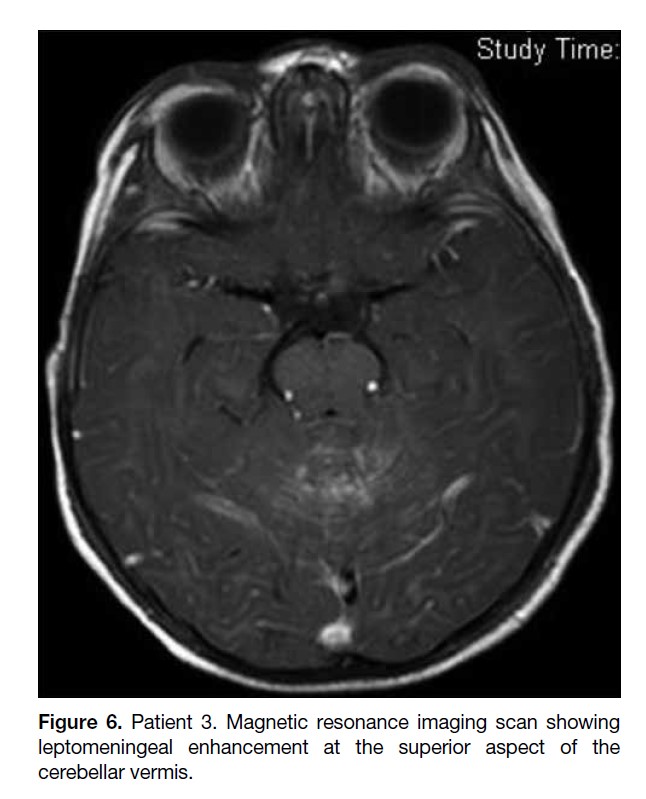
also a cluster of tiny enhancing foci at the superior aspect
of the cerebellar vermis suspicious of leptomeningeal
enhancement (Figure 6). Restricted diffusion was present
along the gyri and subcortical white matter of bilateral
cerebrum and deep grey matter affecting the thalami and
lentiform nuclei (Figure 5c).
Figure 5. Patient 3. Magnetic resonance imaging scans showing (a) T2 and (b) fluid-attenuated inversion recovery high-intensity signals
along the gyri and subcortical white matter of bilateral cerebrum and cerebellum; and (c) restricted diffusion along gyri and subcortical white
matter of bilateral cerebrum and deep grey matter.
Figure 6. Patient 3. Magnetic resonance imaging scan showing
leptomeningeal enhancement at the superior aspect of the
cerebellar vermis.
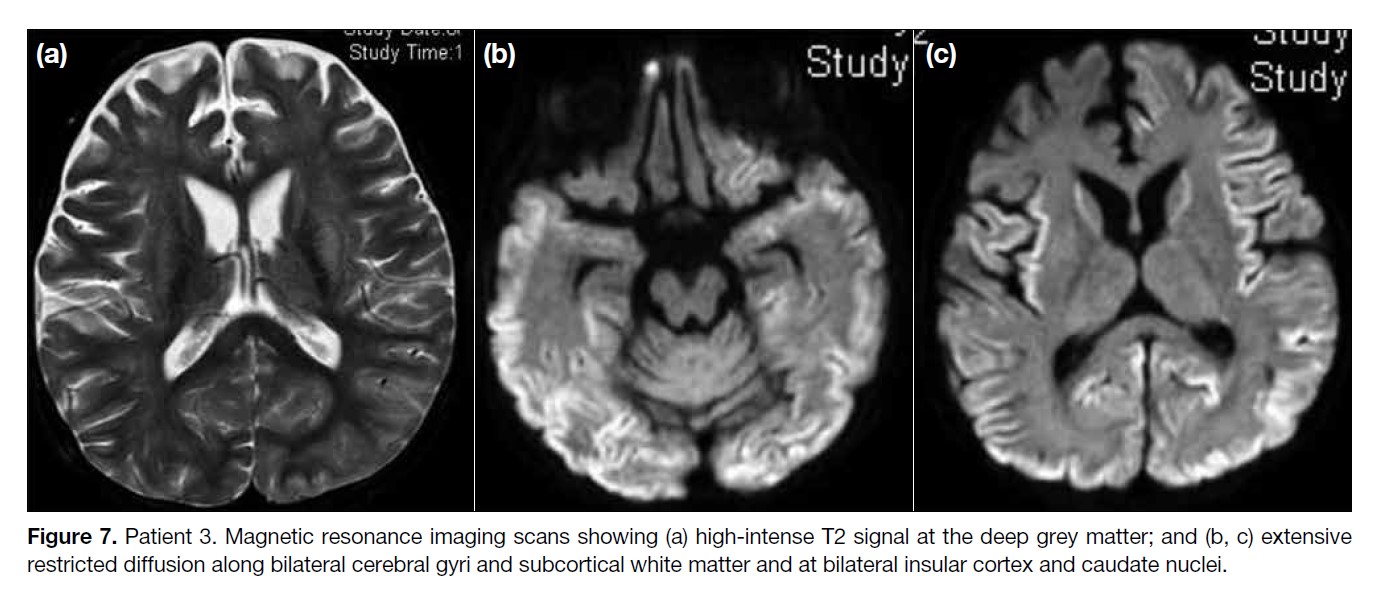
Repeat MRI scan after 2 weeks revealed increased highintensity
T2 signal at the deep grey matter, especially
at the thalami and lentiform nuclei (Figure 7a), with
atrophic changes to the cerebellum. There was partially
resolved contrast enhancement at the periphery and
edges of the white matter lesions. Previously seen
abnormal leptomeningeal enhancement at the superior
aspect of the cerebellar vermis also showed partial
resolution. However, there was more extensive restricted
diffusion along bilateral cerebral gyri and subcortical
white matter and new restricted diffusion at bilateral
insular cortex and caudate nuclei (Figure 7b and c).
Magnetic resonance spectroscopy performed at the same
time showed decreased choline, creatine and N-acetyl
aspartate at the deep grey nuclei and cerebellar lesions
and focal areas of lactate peak.
Figure 7. Patient 3. Magnetic resonance imaging scans showing (a) high-intense T2 signal at the deep grey matter; and (b, c) extensive
restricted diffusion along bilateral cerebral gyri and subcortical white matter and at bilateral insular cortex and caudate nuclei.
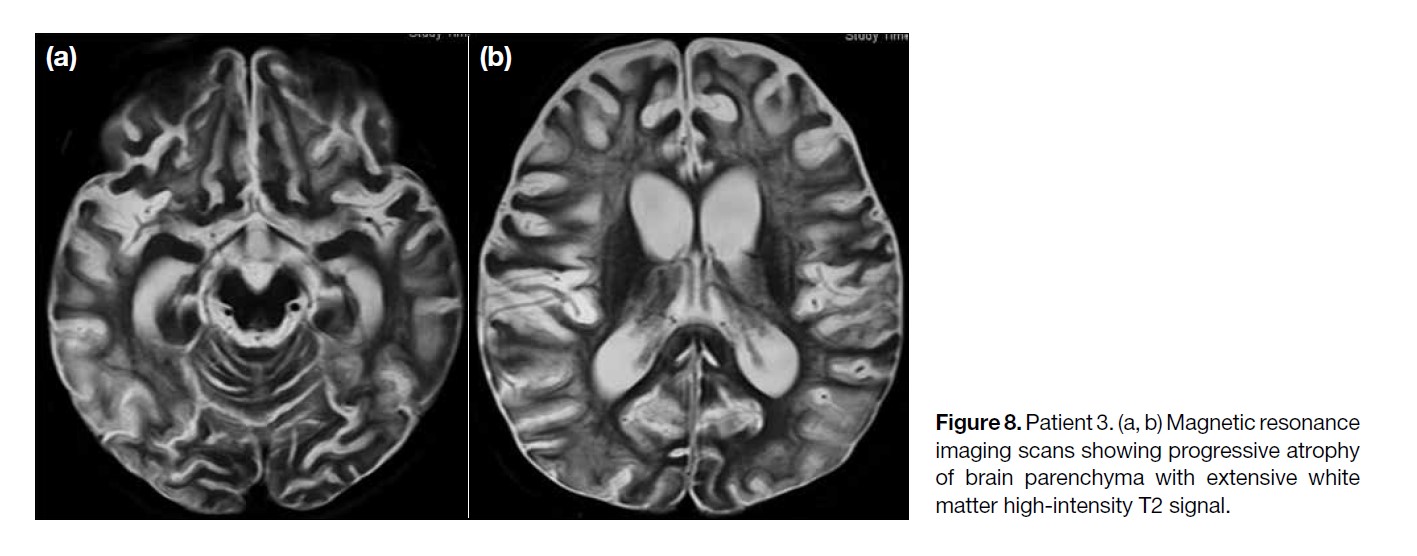
The MRI scan performed 1 month later showed
progressive brain parenchymal loss with extensive
white matter high-intensity T2 signal, also involving the
lentiform nuclei and cerebellum (Figure 8). Progressive
ventriculomegaly was also observed, compatible with
generalised brain parenchymal atrophy.
Figure 8. Patient 3. (a, b) Magnetic resonance
imaging scans showing progressive atrophy
of brain parenchyma with extensive white
matter high-intensity T2 signal.
DISCUSSION
HLH is a progressively fatal disease characterised by
uncontrolled proliferation of activated, non-neoplastic
lymphocytes and macrophages, resulting in phagocytosis
of other blood cells and overproduction of inflammatory
cytokines. It can be divided into primary and secondary
forms. The primary (familial) form of HLH (as in our
case series) is related to genetic abnormalities with an
autosomal recessive mode of inheritance and is usually
diagnosed in the first 2 years of life. The secondary form
is associated with infection, malignancy, and prolonged
immunosuppression.
The clinical and neuroradiological findings are
indistinguishable between the two forms of HLH and
most clinical episodes in the familial form are triggered
by infection. Therefore, any underlying infection or
causative agent should be sought even when suspecting
fHLH.
The diagnosis of HLH is based on established clinical and
laboratory diagnostic criteria, including family history,
fever, splenomegaly, cytopenia of at least two cell lines,
hypertriglyceridaemia, hypofibrinogenaemia, elevated
ferritin, histological evidence of haemophagocytosis in
the bone marrow, CSF, or lymph nodes and detection
of genetic mutations.[1] The neuroradiological findings
are supportive but not sufficiently specific to achieve the
diagnosis.
It is known that the clinical outcome of HLH is poor,
particularly if untreated, but the introduction of specific
treatment protocols (HLH-94 and HLH-2004) has
improved the prognosis. More recently, stem cell or
bone marrow transplantation has become a promising
treatment option for patients with primary HLH.[2]
In addition to sepsis and haemorrhage, central nervous
system (CNS) involvement is another main contributor
to morbidity and mortality in HLH. CNS involvement
is frequent and often progressive but can be treated.
Therefore, the importance of early detection, accurate
evaluation of severity, and monitoring of treatment
response with neuroimaging studies cannot be stressed
enough.
Depending on the patient’s clinical presentation and
neuroimaging findings, other differential diagnoses
may have to be considered, including brain abscess,
metastasis, multiple sclerosis, acute disseminating
encephalomyelitis, lymphoma, malignant glioma and
even child abuse.
In paediatric patients with CNS involvement, the
histopathological findings can be classified by the
microscopic stages of disease, as characterised by
increasing severity[3,4]: stage I shows leptomeningeal
lymphocyte and histiocyte infiltration; stage II represents
additional parenchymal involvement with perivascular
infiltration; and stage III progresses to cerebral tissue
necrosis, gliosis and demyelination in addition to
massive tissue infiltration most significantly affecting
the white matter.
A correlation between the histopathological stage and
CNS imaging findings has been described.[3,4,5] CNS
infiltration typically begins in the meninges, and then
induces perivascular changes.[6] Radiographically, stage I
is represented by leptomeningeal enhancement, as seen
on the initial MRI in one of our patients (Figure 6). The
extent of enhancement in HLH likely correlates with
the degree of meningeal infiltration by lymphocytes and
histiocytes. In previous studies, leptomeningeal contrast
enhancement has also been shown to correlate with
clinical symptoms of meningitis.[4]
Imaging findings for stages II and III include diffuse white
matter changes with variable enhancement, haemorrhage
and restricted diffusion, progressing to parenchymal
infarction, calcifications, and atrophy. Other imaging
findings include delayed myelination, hydrocephalus,
and subdural fluid collections.[3,4] Our patients presented
with a wide spectrum of imaging findings encompassing
different stages.
Enhancing parenchymal lesions have been described in
HLH with CNS involvement, and predominantly affect
the cerebrum and cerebellum.[7,8,9] In one of our cases, a
large right cerebellar enhancing lesion was shown on
MRI. Additionally, nodular or ring-enhancing brain
parenchymal lesions have been reported,[9,10] as seen in
one of our patients (Figure 4c) and is postulated to be
due to the compromised blood-brain barrier associated
with active demyelination.
Ventriculomegaly identified in patients with CNS
involvement in HLH may be due to communicating
hydrocephalus or parenchymal atrophy. In two of our
cases, progressive ventricular enlargement and widening
of the sulcal spaces was likely due to parenchymal
atrophy. In cases of communicating hydrocephalus, the
underlying cause may be a disturbance in CSF drainage
due to leptomeningeal infiltration.
Assessment of restricted diffusion on diffusion-weighted
imaging sequence is useful in cases of HLH. Restricted
diffusion in white matter lesions in HLH is well reported
in the literature,[8] and it was also evident in one of our
patients. This is postulated to be due to neuronal loss
along with cytotoxic oedema and demyelination in the
acute phase of the disease.[3]
Overall, the severity of CNS involvement in HLH
is variable and related to the duration of illness. The
neuroimaging findings correlate with clinical evolution
and are useful to monitor disease progression or
regression. Imaging findings should be correlated
with clinical symptoms in addition to laboratory and
pathological findings in order to narrow the differential
diagnosis and provide timely diagnosis of patients with
HLH.
CONCLUSION
fHLH is a rare hereditary disease and can present with
a wide range of neuroradiological findings. Despite the
typically non-specific clinical presentation, awareness of
this entity and familiarity with the spectrum of imaging
findings may facilitate early diagnosis and timely
commencement of appropriate treatment to modify and
improve the disease course.
REFERENCES
1. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166:95-109. Crossref
2. Hallahan AR, Carpenter PA, O'Gorman-Hughes DW, Vowels MR, Marshall GM. Hemophagocytic lymphohistiocytosis in children. J Paediatr Child Health. 1999;35:55-9. Crossref
3. Chung TW. CNS involvement in hemophagocytic lymphohistiocytosis: CT and MR findings. Korean J Radiol. 2007;8:78-81. Crossref
4. Anderson TL, Carr CM, Kaufmann TJ. Central nervous system imaging findings of hemophagocytic syndrome. Clin Imaging. 2015;39:1090-4. Crossref
5. Rooms L, Fitzgerald N, McClain KL. Hemophagocytic lymphohistiocytosis masquerading as child abuse: presentation of three cases and review of central nervous system findings in hemophagocytic lymphohistiocytosis. Pediatrics. 2003;111(5 Pt 1):e636-40. Crossref
6. Kim MM, Yum MS, Choi HW, Ko TS, Im HJ, Seo JJ, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol. 2012;47:273-80. Crossref
7. Shinoda J, Murase S, Takenaka K, Sakai N. Isolated central nervous system hemophagocytic lymphohistiocytosis: case report. Neurosurgery. 2005;56:E187-90. Crossref
8. Ozgen B, Karli-Oguz K, Sarikaya B, Tavil B, Gurgey A. Diffusion-weighted cranial MR imaging findings in a patient with hemophagocytic syndrome. AJNR Am J Neuroradiol. 2006;27:1312-4.
9. Goo HW, Weon YC. A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr Radiol. 2007;37:1110-7. Crossref
10. Akima M, Sumi SM. Neuropathology of familial erythrocytic lymphohistiocytosis: six cases and review of the literature. Hum Pathol. 1984;15:161-8. Crossref