Familial Amyloidotic Polyneuropathy with Leptomeningeal and Cardiac Involvement in a Patient with Gly73Glu Transthyretin Gene Mutation — Non-invasive Diagnostic Approach with Multimodality Imaging Findings: a Case Report
CASE REPORT
Familial Amyloidotic Polyneuropathy with Leptomeningeal and Cardiac Involvement in a Patient with Gly73Glu Transthyretin Gene Mutation — Non-invasive Diagnostic Approach with Multimodality Imaging Findings: a Case Report
BCK Chow1, SSM Lo2, JCY Lee1, JB Chiang1, HF Chan3, CB Ho3, LT Szeto4, KW Tang1
1 Department of Radiology and Imaging, Queen Elizabeth Hospital, Hong Kong
2 Scanning Department, St. Teresa’s Hospital, Hong Kong
3 Department of Medicine, Queen Elizabeth Hospital, Hong Kong
4 Department of Nuclear Medicine, Queen Elizabeth Hospital, Hong Kong
Correspondence: Dr BCK Chow, Department of Radiology and Imaging, Queen Elizabeth Hospital, Hong Kong. Email: chowbck@gmail.com
Submitted: 29 Oct 2020; Accepted: 19 Jan 2021.
Contributors: BCKC, SSML and JCYL designed the study. All authors acquired the data. BCKC analysed the data and drafted the manuscript. SSML, JCYL, JBC, HFC, CBH, LTS and KWT critically revised the manuscript for important intellectual content.
Conflicts of Interest: All authors have disclosed no conflicts of interest.
Funding/Support: This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data Availability: All data generated or analysed during the present study are included in this published article.
Ethics Approval: The patients were treated in accordance with the tenets of the Declaration of Helsinki. Verbal informed consent for all treatments and procedures was obtained from the patient and her husband.
CASE REPORT
In April 2020, a 47-year-old woman presented with
peripheral upper and lower limb numbness and recurrent
headaches, and a history of transient expressive
dysphasia with spontaneous recovery 6 years previously.
She also developed gradual cognitive impairment and
bilateral sensorineural hearing loss after that. Extensive
blood tests and imaging including echocardiogram were
all unremarkable. Nerve conduction testing revealed
demyelinating sensorimotor polyneuropathy of the
lower limbs. Three months previously the patient was
admitted with rapid deterioration in her physical and
mental condition, as well as lower limb oedema. Chest
radiograph showed new bilateral pleural effusions.
Magnetic resonance imaging (MRI) brain studies
revealed diffuse leptomeningeal enhancement mainly
in the central skull base cisterns and bilateral posterior cranial fossa (Figure 1a and b) and diffuse superficial
haemosiderosis on susceptibility-weighted imaging
(Figure 1c). Subsequent development of a left frontal
lobe intracerebral haemorrhage was noted on follow-up
images (Figure 1d). Echocardiogram revealed a
moderately impaired left ventricular ejection function
of 35% and left ventricular hypertrophy with infiltrative
features (Figure 2). Blood test for serum free light
chain level, as well as urine for proteins were not
elevated. Subsequent cardiac MRI (Figures 3 and 4)
and technetium-99m pyrophosphate scan (Figure 5)
confirmed transthyretin (TTR) amyloidosis. Genetic
study indicated a pathological TTR gene (p.Gly73Glu
in exon 3). Retrospective review of her family history
revealed that her father had died from a cerebral
vascular accident in his thirties, and her mother from
an unknown neurological condition at an early age. The diagnosis of hereditary TTR amyloidosis with
cardiac and neurological involvement was made. The
patient was referred for detailed evaluation and potential
pharmacological treatment of familial amyloidotic
polyneuropathy.
Figure 1. Magnetic resonance imaging (MRI) images of the brain. T1-weighted post-gadolinium axial images of the brain at the level of (a) lateral ventricles and (b) the pons demonstrating diffuse leptomeningeal enhancement involving bilateral cerebral sulci and perimesencephalic region. (c) Susceptibility-weighted image showing diffuse superficial haemosiderosis. (d) T1-weighted axial images of follow-up MRI of the brain 1 month later, showing T1-weighted hyperintense intracerebral haematoma in the left inferior frontal lobe.
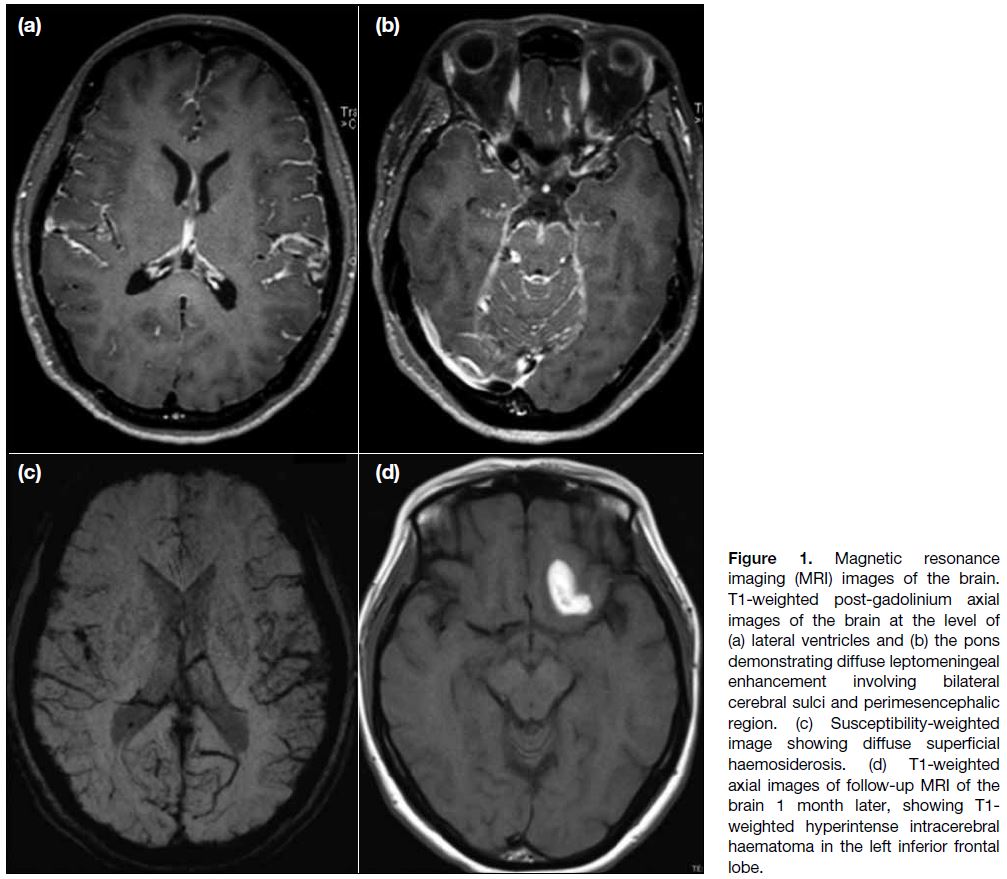
Figure 2. (a) Echocardiogram in parasternal long axis view showing marked increase in left ventricular wall thickness. (b) Strain image
showing decreased global longitudinal strain with apical sparing.
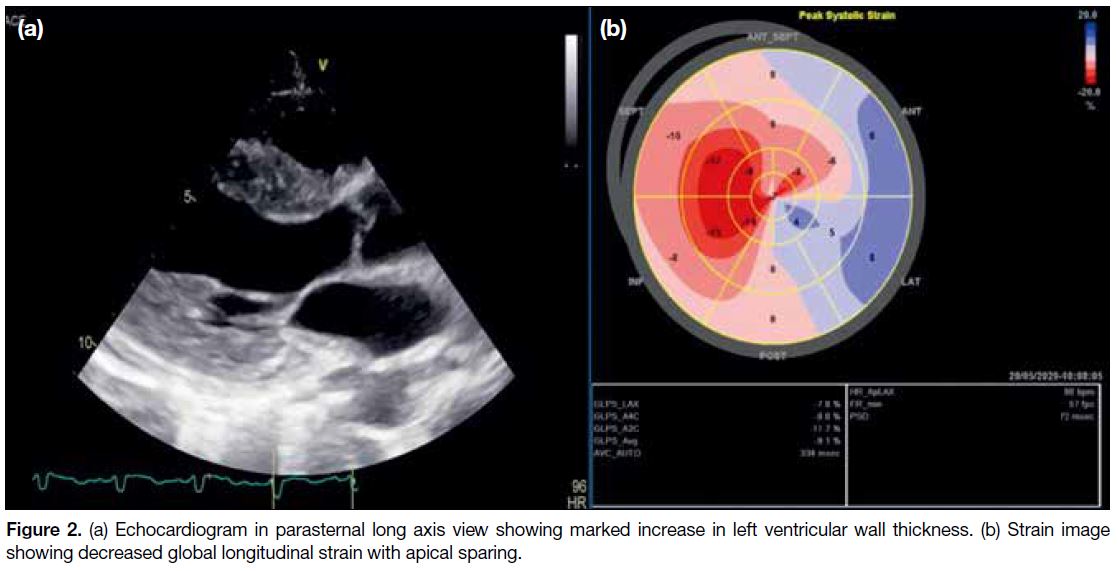
Figure 3. Cardiac magnetic resonance imaging late gadolinium-enhanced short axis images of the left ventricle at the (a) basal, (b) mid and
(c) apical levels demonstrating global subendocardial delayed enhancement not following coronary territories.
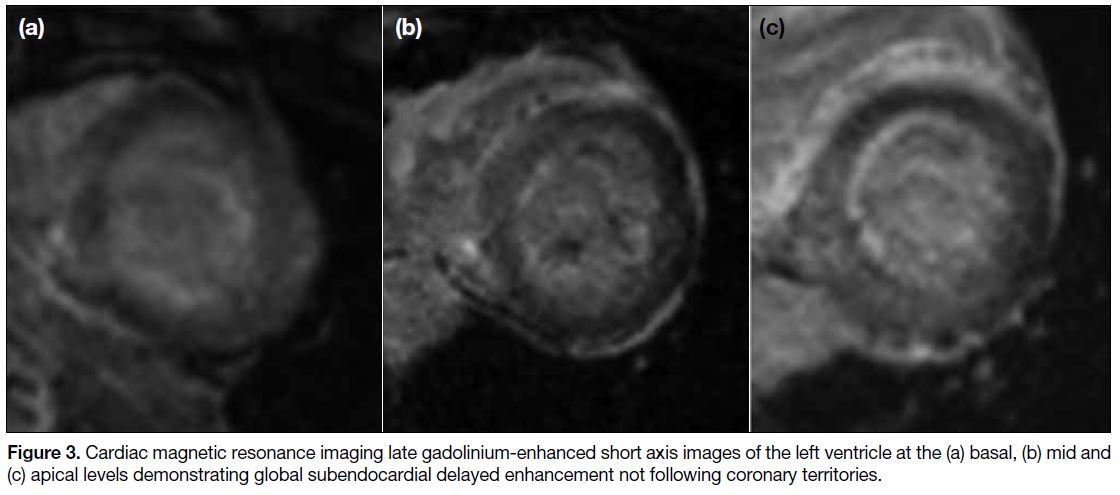
Figure 4. Cardiac magnetic resonance imaging native T1 mapping
showing marked elevation of T1 value of the myocardium,
measuring 1273 ms (normal myocardial T1 mapping value in our
centre: 1019 ± 41 ms).
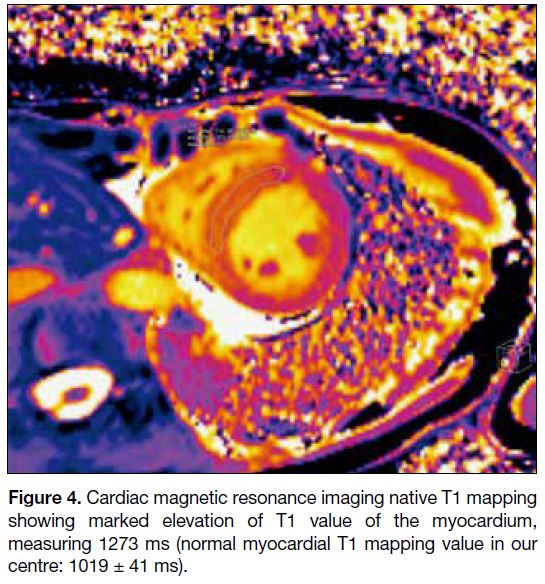
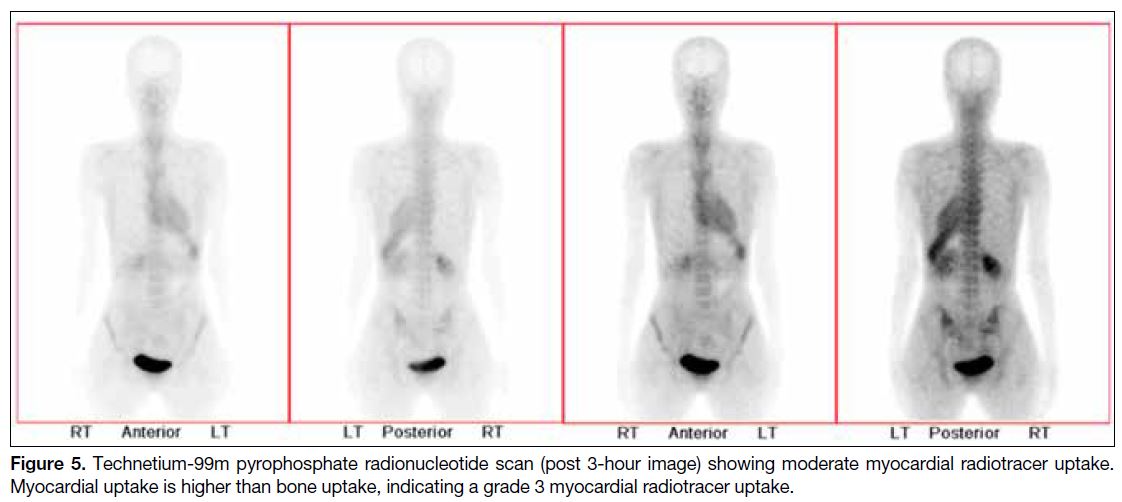
Figure 5. Technetium-99m pyrophosphate radionucleotide scan (post 3-hour image) showing moderate myocardial radiotracer uptake. Myocardial uptake is higher than bone uptake, indicating a grade 3 myocardial radiotracer uptake.
DISCUSSION
Hereditary TTR amyloidosis is an exceptionally rare
disease. Few cases of hereditary TTR amyloidosis with
cardiac involvement or peripheral neuropathy from
Val142Ala[1] or Ala117Ser[2] mutations have been reported
in Hong Kong. There have been some cases reported
from France[3] and Sweden[4], but this is the first in Asia
with leptomeningeal involvement of amyloidosis from a
Gly73Glu TTR gene mutation.
Familial amyloidotic polyneuropathy is a familial
disease characterised by accumulation of amyloid
fibrillar proteins in organs and peripheral nerves. Onset
is usually between the ages of 30 and 40 years. Systemic
involvement includes sensorimotor polyneuropathy,
autonomic dysfunction, and cardiac, renal, and hepatic
involvement.
TTR amyloidosis is the most common familial
amyloidosis. The genetic mutation at the TTR
amyloidosis gene causes destabilisation and dissociation
of the TTR protein, leading to misfolded monomers
that ultimately self-assemble to form amyloid fibrils.
There are more than 100 reported mutations of the TTR
gene, with Val30Met being the most common. Some forms, including the Gly73Glu mutation, will lead to
a preferential involvement of the leptomeninges and
meningovascular walls, as well as amyloid-derived
vitreous opacity, thus they are termed leptomeningeal or
oculoleptomeningeal amyloidosis.
Clinically these patients present with manifestations
including headache, dementia, ataxia, spastic paralysis
or convulsion.[5] MRI is the most sensitive modality to
identify the leptomeningeal abnormalities, demonstrating intermediate T1-weighted signal intensity and contrast
enhancement of the leptomeninges along the Sylvian
fissures, cerebral sulci, cisterns and surface of the
brainstem, as in our case, and also along the surface of
the cerebellum and spinal cord. Due to the abnormal
cerebral and leptomeningeal vessels, these patients
are prone to intracerebral, subarachnoid, or subdural
haemorrhages.
There are multiple differentials for diffuse leptomeningeal enhancement. Nodular leptomeningeal
thickening is usually seen in leptomeningeal
carcinomatosis. Tuberculous meningitis and
neurosarcoidosis can both demonstrate smooth or
nodular leptomeningeal enhancement although they are
predominantly located in the vicinity of basal cisterns. It
can sometimes be difficult to differentiate various causes
of leptomeningeal disease. Compatible clinical features
should raise prompt consideration of a diagnosis of familial cerebral amyloid angiopathies.
The pathogenesis of sporadic-type cerebral amyloid
angiopathy is vastly different to that of the familial type.
The sporadic type more commonly affects the elderly
people. Unlike familial cerebral amyloid angiopathy, the
sporadic form is characterised by progressive amyloid-β
protein deposition on the walls of small- to medium-sized
arteries, arterioles and capillaries in the cerebral
and cerebellar cortices, with vessel wall thickening,
endothelial dysfunction and a loss of compliance leading
to fragile vessels. This causes intracranial macro- and
micro-haemorrhages. On MRI, gradient echo sequences
or susceptibility-weighted imaging are helpful to depict
these macro- and micro-bleeds that are usually distributed
in a lobar predilection. Other non-specific findings
include cerebral atrophy and cerebral white matter signal
changes. There is seldom extensive leptomeningeal
deposition or enhancement in the sporadic type.
Cardiac involvement is also common in familial
amyloidosis, and patients usually present with heart
failure or refractory arrhythmia. Endomyocardial biopsy
has historically been the gold standard for a definitive
diagnosis of cardiac amyloidosis. Recent study suggests
that suspicious cardiac MRI findings with a grade 2 or 3
myocardial radiotracer uptake on bone scintigraphy in a
patient with no monoclonal protein in serum or urine has
specificity and positive predictive value of 100% in the
diagnosis of TTR cardiac amyloidosis.[6] This obviates the
need for endomyocardial biopsy in some patients.
There are several pharmacological interventions that can
be applied to prevent the formation of amyloid proteins.
They include suppression of TTR synthesis (patisiran,
inotersen), stabilisation of TTR to prevent misfolding
into amyloid proteins (tafamidis, diflunisal), as well as
TTR fibril degradation and absorption (doxycycline-tauroursodeoxycholic
acid, monoclonal anti-serum
amyloid protein antibody).[7] Patients may also benefit
from liver transplantation or a combined heart and liver
transplant, with potential long-term histopathologic
regression of amyloid deposits. Prognosis is variable,
depending on the TTR variants, age, nutritional status
and the severity of neuropathy and cardiac amyloid
involvement. A multidisciplinary approach is often key
to successful management of patients with hereditary
amyloidosis.
In conclusion, the finding of a constellation of unique neurological and cardiac findings in this patient utilising
various imaging modalities enabled us to diagnose this
uncommon multisystem disease whose initial clinical
presentation is often non-specific.
REFERENCES
1. Wong CW, Ng WY, So KL, Chan YH, Yip SF, Mak CM. A
rare variant of transthyretin-related amyloidosis associated with
exclusive cardiomyopathy in a Hong Kong Chinese patient. J
Cardiol Cases. 2018;18:185-8. Crossref
2. Ho MH, Yip PL, Ho CB, Wong FC, Chen SP, Wong CY. Non-biopsy
diagnosis of hereditary transthyretin amyloidosis presented
with cardiomyopathy and peripheral neuropathy in a Chinese man
in Hong Kong. Int J Rare Dis Disord. 2020;3:18. Crossref
3. Ellie E, Camou F, Vital A, Rummens C, Grateau G, Delpech M,
et al. Recurrent subarachnoid hemorrhage associated with a new
transthyretin variant (Gly53Glu). Neurology. 2001;57:135-7. Crossref
4. Holmgren G, Hellman U, Anan I, Lundgren HE, Jonasson J,
Stafberg C, et al. Cardiomyopathy in Swedish patients with
the Gly53Glu and His88Arg transthyretin variants. Amyloid.
2005;12:184-8. Crossref
5. Nakamura M, Yamashita T, Ueda M, Obayashi K, Sato T, Ikeda T, et al. Neuroradiologic and clinicopathologic features of
oculoleptomeningeal type amyloidosis. Neurology. 2005;65:1051-6. Crossref
6. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-12. Crossref
7. Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging diseasemodifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20:163-78. Crossref