Aceruloplasminemia with Neurodegenerative Condition: A Case Report
CASE REPORT
Hong Kong J Radiol 2025 Jun;28(2):e107-10 | Epub 19 June 2025
Aceruloplasminemia with Neurodegenerative Condition: A Case Report
CK Li, CY Lau, KH Chin, CY Chu
Department of Radiology, Pamela Youde Nethersole Eastern Hospital, Hong Kong SAR, China
Correspondence: Dr CK Li, Department of Radiology, Pamela Youde Nethersole Eastern Hospital, Hong Kong SAR, China. Email: lck340@ha.org.hk
Submitted: 4 May 2024; Accepted: 3 September 2024.
Contributors: CKL designed the study, acquired and analysed the data, and drafted the manuscript. CYL, KHC and CYC critically revised the manuscript for important intellectual content. All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have disclosed no conflicts of interest.
Funding/Support: This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data Availability: All data generated or analysed during the present study are available from the corresponding author on reasonable request.
Ethics Approval: The study was approved by the Central Institutional Review Board of Hospital Authority, Hong Kong (Ref No.: IRB-2024-245). The patient was treated in accordance with the tenets of the Declaration of Helsinki. Informed verbal consent was obtained from the patient’s first-degree relative for the publication of this case report, including the accompanying images.
CASE PRESENTATION
A 68-year-old Chinese woman presented to the Accident
and Emergency Department of our institution in March
2023 with confusion, gait instability, and a history of
falls. She had experienced a rapid decline in mobility and
motivation, rendering her homebound since December
2022. Her medical history revealed repetitive behaviour
spanning over a decade, alongside co-morbidities such
as diabetes mellitus and mild anaemia since 2007.
Neither the patient nor her relatives reported seizures or
loss of consciousness. Physical examination showed no
focal neurological deficits. Dementia evaluation by the
Montreal Cognitive Assessment test yielded a score of 2
out of 30, indicating a high clinical suspicion.
Non-contrast computed tomography (CT) of the brain
was unremarkable with known chronic ventriculomegaly
as the only notable finding. Subsequent contrast-enhanced
magnetic resonance imaging (MRI) showed
extensive symmetrical blooming artefacts in various
deep grey matter areas on the susceptibility-weighted
imaging (SWI) sequence, including the bilateral caudate nuclei, lentiform nuclei, thalami, red nuclei, substantia
nigra, and bilateral dentate nuclei of the cerebellum.
Diffuse gyriform-like blooming artefacts were
observed outlining the surfaces of the cerebrum and
cerebellum (Figure 1). These MRI findings suggested
significant mineral deposition, raising suspicion of
aceruloplasminemia and other differential diagnoses such
as other neurodegeneration with brain iron accumulation.
In view of the suspected iron accumulation, contrast-enhanced
CT of the abdomen and the pelvis, as well
as MRI of the liver and the heart, were performed.
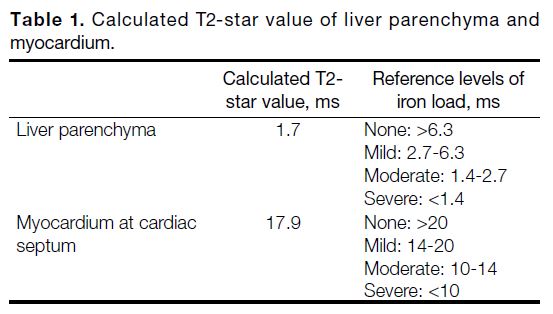
The CT scan revealed diffuse hyperattenuation of the
liver parenchyma, while MRI showed evidence of iron
overload in both the liver parenchyma and myocardium
(Table 1).
Figure 1. Brain magnetic resonance
imaging (3T). Axial susceptibility-weighted
sequence shows extensive symmetrical
blooming artefacts in deep grey matter
areas, including (a) bilateral caudate nuclei
(yellow arrows), lentiform nuclei (white
arrows), thalami (red stars); (b) red nuclei
(yellow circles), substantia nigra (orange
arrows); and (c) bilateral dentate nuclei of
the cerebellum (red arrows). (d, e) Diffuse
gyriform-like blooming artefacts outlining
the surfaces of the cerebrum and cerebellum
(yellow arrowheads).
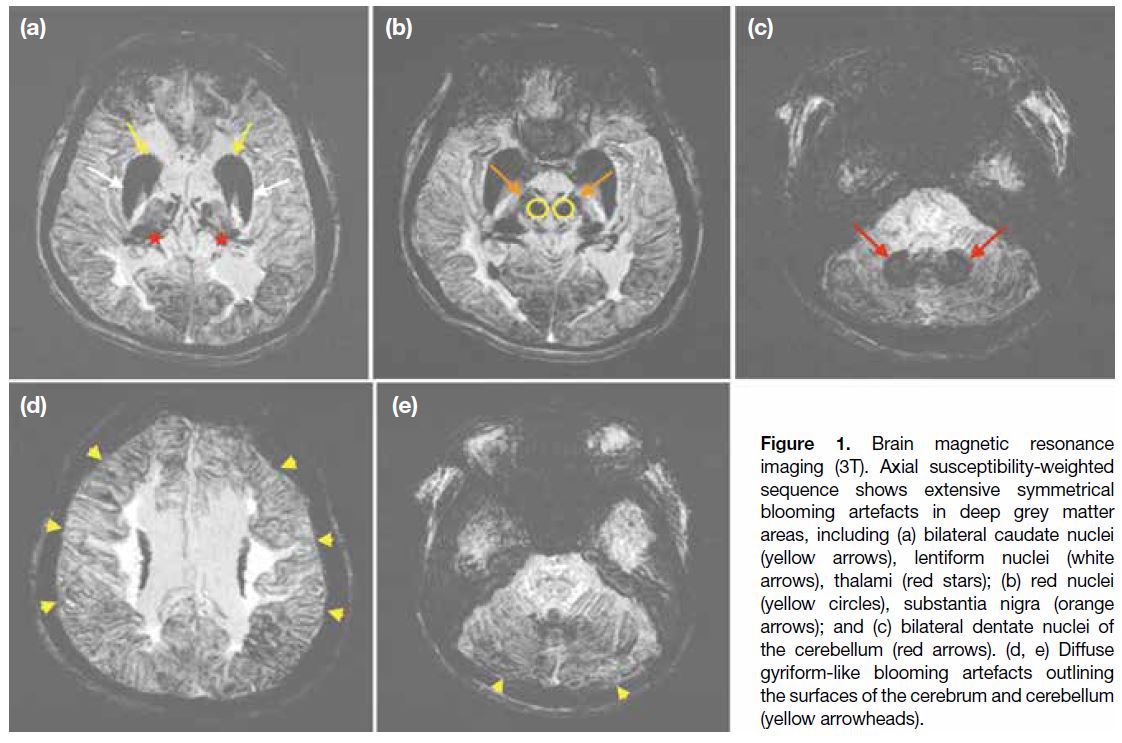
Table 1. Calculated T2-star value of liver parenchyma and myocardium.
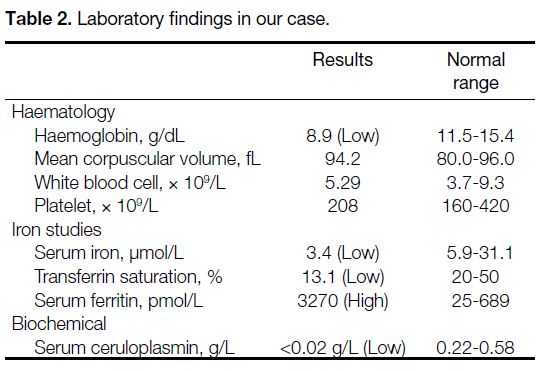
Biochemically, the patient exhibited a markedly low
ceruloplasmin level of under 0.02 g/L (normal range = 0.22-0.58), an elevated ferritin level of 3270 pmol/L
(normal range = 25-689), and a low iron saturation of
13.1% (Table 2). She also had a history of chronic mild
anaemia for at least a decade, with haemoglobin levels ranging from 10.4 g/dL in April 2013 to 8.9 g/dL in
March 2023. Genetic testing subsequently identified a
pathogenic variant of the ceruloplasmin gene, confirming
the diagnosis of aceruloplasminemia.
Table 2. Laboratory findings in our case.
The patient and her relative were counselled about the
definitive diagnosis, and the features of the disease
were explained. No specific treatment was prescribed
for aceruloplasminemia due to chronic neurological
symptoms and impaired cognitive function. The patient
continued to receive holistic care in a residential elderly
care home, with monitoring for her diabetes. Genetic
testing was also offered to her first-degree relatives.
DISCUSSION
Aceruloplasminemia is a rare autosomal recessive
disorder characterised by the absence or dysfunction
of ceruloplasmin with consequent iron accumulation
in various tissues and organs, leading to a spectrum of
neurological and systemic manifestations.[1] Our case
illustrates the importance of recognising the clinical and
radiological features of aceruloplasminemia to facilitate
accurate diagnosis and management.
Aceruloplasminemia was first documented in 1987
by Miyajima et al[2] in a 52-year-old woman with
blepharospasm, retinal degeneration, and diabetes
mellitus. The estimated prevalence is approximately 1 in
2,000,000 population among Japanese individuals born
from non-consanguineous marriages.[3] Nonetheless, this
estimation is region-specific and may not be applicable
to other populations.[4] Clinical manifestations leading to
diagnosis by neurologists include cerebellar signs such
as dysarthria, trunk and limb ataxia, and involuntary
movements including dystonia, chorea, and tremors.
Symptoms may vary widely among individuals and may
overlap with other neurological or metabolic disorders.[5]
To understand the pathophysiology of
aceruloplasminemia, two distinct isoforms of
ceruloplasmin are produced via alternative splicing
in exons 19 and 20, resulting in a soluble form in
plasma and a glycosylphosphatidylinositol-anchored
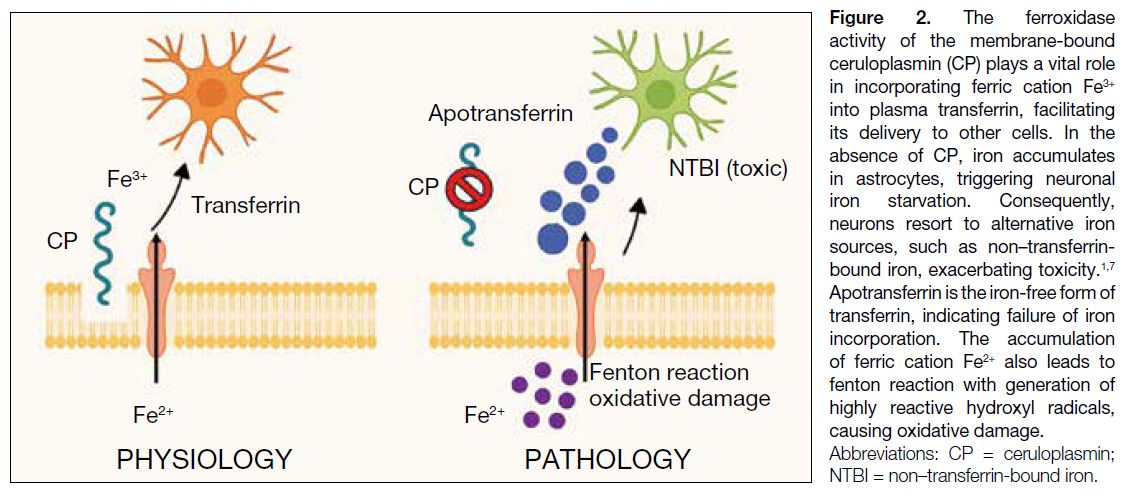
membrane form.[6] The ferroxidase activity of the
membrane-bound ceruloplasmin plays a vital role for
incorporating ferric cation Fe3+ into plasma transferrin,
facilitating its delivery to other cells via transferrin
receptor 1. In the absence of ceruloplasmin, iron
initially accumulates in astrocytes, triggering neuronal
iron starvation. Consequently, neurons resort to
alternative iron sources such as non–transferrin-bound
iron, exacerbating toxicity (Figure 2).[1] [7]
Figure 2. The ferroxidase
activity of the membrane-bound
ceruloplasmin (CP) plays a vital role
in incorporating ferric cation Fe3+
into plasma transferrin, facilitating
its delivery to other cells. In the
absence of CP, iron accumulates
in astrocytes, triggering neuronal
iron starvation. Consequently,
neurons resort to alternative iron
sources, such as non–transferrin-bound
iron, exacerbating toxicity.1,7
Apotransferrin is the iron-free form of
transferrin, indicating failure of iron
incorporation. The accumulation
of ferric cation Fe2+ also leads to
fenton reaction with generation of
highly reactive hydroxyl radicals,
causing oxidative damage.
The hallmark radiological feature of aceruloplasminemia
manifests as symmetric blooming artefacts on SWI,
attributable to iron accumulation in the brain. Typically, this involves regions such as the basal ganglia and
thalamus, cerebral cortex and dentate nuclei of the
cerebellum.[8] Aceruloplasminemia stands out as the sole
recognised disorder featuring both cerebral and systemic
manifestations of iron accumulation.[9] As in our patient,
cardiac and hepatic iron overload may also occur. Hepatic
iron overload often presents with hyperattenuation of
the liver parenchyma on CT scans and is quantitatively
assessed via MRI dedicated to evaluating iron overload
in the liver. Nonetheless, liver iron accumulation seldom
leads to clinical manifestations such as cirrhosis or liver
failure.[10] Iron deposition in other organs, including the
heart, pancreas, and other endocrine glands, has been
documented and can be evaluated by MRI.[7]
The neurological manifestations of aceruloplasminemia
are heterogeneous and often progressive. In our patient,
initial symptoms such as confusion, gait instability, and
falls were consistent with those commonly reported in the
literature. Documented neurological features included
behavioural changes or psychiatric manifestations,
cognitive impairment, extrapyramidal signs, cerebellar
signs, and involuntary movements.[5] Another classic
clinical manifestation is diabetes mellitus, typically
presenting in the fourth to sixth decades of life in
individuals without classic risk factors or need for
insulin treatment.[11] The mechanism underlying the
development of diabetes mellitus in aceruloplasminemia
remains poorly understood, although iron accumulation
is noted predominantly in exocrine rather than
endocrine pancreatic cells.[12] Some studies suggest
that the clinical triad of aceruloplasminemia may comprise neurodegeneration, diabetes mellitus, and
retinal degeneration.[13] [14] Nonetheless retinopathy is less
frequently observed in non-Japanese case series, and
its direct association with aceruloplasminemia remains
uncertain.[13] [14]
Biochemically, the first detectable parameters of
aceruloplasminemia, as indicated by all major case series
including our own, encompass mild microcytic anaemia,
low transferrin saturation, and hyperserotonaemia. This
biochemical triad holds crucial diagnostic significance
long before other clinical manifestations emerge. Serum
ceruloplasmin is typically undetectable or markedly
reduced and serves as an important diagnostic parameter.
Although mild microcytic anaemia often emerges as the
earliest biochemical sign of aceruloplasminemia,[5] [10] it
rarely leads to diagnosis at the early pre-symptomatic
stage. By integrating biochemical studies with
radiological and clinical manifestations, the exclusion of
other differential neurodegenerative diseases becomes
more manageable. As in our case, genetic testing
provides definitive evidence to confirm the diagnosis of
aceruloplasminemia and enables genetic counselling and
family screening for at-risk individuals.
Treatment of aceruloplasminemia primarily involves
iron-chelating agents; however, their effectiveness
in reducing brain iron and alleviating neurological
symptoms remains uncertain. Currently, there is no
convincing evidence supporting the clinical benefits of
iron removal therapy. Phlebotomy, another treatment
option, is also considered suboptimal. Alternative
strategies focus on preventing oxidative tissue damage,
such as administering vitamin E or zinc sulphate.[10]
Timely diagnosis and treatment are paramount to prevent
irreversible neurological complications.[7]
CONCLUSION
Aceruloplasminemia is difficult to diagnose and requires
a high level of awareness of its clinical features,
biochemical parameters, and radiological findings.
The biochemical triad of mild anaemia, low transferrin
saturation, and hyperserotonaemia serves as a key
diagnostic indicator when no alternative explanation is
evident. The condition should be considered in patients
who present with mild microcytic anaemia, early-onset
diabetes mellitus, and unexplained liver iron overload. In
later stages, adult-onset neurological dysfunction, such as behavioural changes, psychiatric disturbances, as well
as cerebellar and extrapyramidal signs, become apparent.
Corresponding MRI findings often reveal symmetrical
hypointensity in the basal ganglia and thalamus, cerebral
cortex and dentate nuclei of cerebellum in T2 and T2-star
sequences, along with a pronounced blooming artifact in
SWI. Prompt diagnosis is crucial to prevent irreversible
neurological complications.
REFERENCES
1. Fasano A, Colosimo C, Miyajima H, Tonali PA, Re TJ,
Bentivoglio AR. Aceruloplasminemia: a novel mutation in a family
with marked phenotypic variability. Mov Disord. 2008;23:751-5. Crossref
2. Miyajima H, Nishimura Y, Mimguchi K, Sakamoto M, Shimizu T,
Honda N. Familial apoceruloplasmin deficiency associated with
blepharospasm and retinal degeneration. Neurology. 1987;37:761-7. Crossref
3. Miyajima H, Kohno S, Takahashi Y, Yonekawa O, Kanno T.
Estimation of the gene frequency of aceruloplasminemia in Japan.
Neurology. 1999;53:617-9. Crossref
4. Yamamura A, Kikukawa Y, Tokunaga K, Miyagawa E, Endo S,
Miyake H, et al. Pancytopenia and myelodysplastic changes in
aceruloplasminemia: a case with a novel pathogenic variant in the
ceruloplasmin gene. Intern Med. 2018;57:1905-10. Crossref
5. Miyajima H, Hosoi Y. Aceruloplasminemia. In: Adam MP,
Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al,
editors. GeneReviews® [Internet]. Seattle (WA): University of
Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1493/". Accessed 4 May 2024.
6. Patel BN, Dunn RJ, David S. Alternative RNA splicing generates
a glycosylphosphatidylinositol-anchored form of ceruloplasmin in
mammalian brain. J Biol Chem. 2000;275:4305-10. Crossref
7. Marchi G, Busti F, Lira Zidanes A, Castagna A, Girelli D.
Aceruloplasminemia: a severe neurodegenerative disorder
deserving an early diagnosis. Front Neurosci. 2019;13:325. Crossref
8. Grisoli M, Piperno A, Chiapparini L, Mariani R, Savoiardo M. MR
imaging of cerebral cortical involvement in aceruloplasminemia.
AJNR Am J Neuroradiol. 2005;26:657-61.
9. Touarsa F, Ali Mohamed D, Onka B, Rostoum S, Ech-Cherif
El Kettani N, Fikri M, et al. Brain iron accumulation on MRI
revealing aceruloplasminemia: a rare cause of simultaneous brain
and systemic iron overload. BJR Case Rep. 2022;8:20220035. Crossref
10. Pelucchi S, Mariani R, Ravasi G, Pelloni I, Marano M, Tremolizzo L,
et al. Phenotypic heterogeneity in seven Italian cases of
aceruloplasminemia. Parkinsonism Relat Disord. 2018;51:36-42. Crossref
11. Vroegindeweij LH, Langendonk JG, Langeveld M, Hoogendoorn M,
Kievit AJ, Di Raimondo D, et al. New insights in the neurological
phenotype of aceruloplasminemia in Caucasian patients.
Parkinsonism Relat Disord. 2017;36:33-40. Crossref
12. Kato T, Daimon M, Kawanami T, Ikezawa Y, Sasaki H, Maeda K.
Islet changes in hereditary ceruloplasmin deficiency. Hum Pathol.
1997;28:499-502. Crossref
13. Miyajima H, Takahashi Y, Kono S. Aceruloplasminemia, an
inherited disorder of iron metabolism. Biometals. 2003;16:205-13. Crossref
14. McNeill A, Pandolfo M, Kuhn J, Shang H, Miyajima H. The
neurological presentation of ceruloplasmin gene mutations. Eur
Neurol. 2008;60:200-5. Crossref